International
AbbVie 2022: Still immune
AbbVie may have doubled in size since the Allergan acquisition, but nearly half the company’s revenue, and a large chunk of its potential future growth,…
Share this:


AbbVie may have doubled in size since the Allergan acquisition, but nearly half the company’s revenue, and a large chunk of its potential future growth, are still coming from a familiar place.
By Joshua Slatko • josh.slatko@medadnews.com
1 North Waukegan Road
North Chicago, IL 60064-6400
847-932-7900 • abbvie.com
| Financial Performance | ||||
| 2021 | 2020 | 1H 2022 | 1H 2021 | |
| Revenue | $56,197 | $45,804 | $28,121 | $26,969 |
| Net income | $11,549 | $4,622 | $5,421 | $4,324 |
| Diluted EPS | $6.45 | $2.72 | $3.03 | $2.41 |
| R&D expense | $7,084 | $6,557 | $3,106 | $3,434 |
| All figures are in millions of dollars, except EPS. | ||||
Best-selling products
All sales are in millions of dollars.
* – Allergan product revenue after acquisition closing date of May 8, 2020.
2021 sales
- Humira $20,694
- Imbruvica $5,408
- Skyrizi $2,939
- Botox Therapeutic $2,451
- Botox Cosmetic $2,232
- Venclexta $1,820
- Vraylar $1,728
- Mavyret $1,710
- Rinvoq $1,651
- Juvederm Collection $1,535
- Restasis $1,290
- Creon $1,191
- Linzess $1,038
- Lupron $783
- Synthroid $767
- Lumigan $579
- Ubrelvy $552
- Alphagan $529
- Duodopa $511
1H 2022 sales
- Humira $10,099
- Imbruvica $2,318
- Skyrizi $2,192
- Botox Cosmetic $1,336
- Botox Therapeutic $1,292
- Rinvoq $1,057
- Venclexta $978
- Vraylar $919
- Mavyret $778
- Juvederm Collection $754
- Creon $605
- Linzess $495
- Restasis $414
- Ubrelvy $323
- Lumigan $270
Outcomes Creativity Index Score: 13
- Manny Awards — N/A
- Cannes Lions — N/A
- Clio Health — N/A
- Creative Floor Awards — 13
- London International Awards – N/A
- MM+M Awards — N/A
- One Show — N/A

CEO and Chairman Richard Gonzalez
Even after the vast Allergan acquisition shook up the company’s product portfolio two years ago, AbbVie still looks a lot like an immunology company. Humira remains, well, Humira, a $20 billion gorilla in the room which is still the world’s best-selling pharmaceutical that is not a COVID vaccine. And the two fastest-growing brands in AbbVie’s stable are, you guessed it, immunology products – Skyrizi (nearly doubled sales in 2021) and Rinvoq (more than doubled sales in 2021), both of which have enjoyed multiple new approvals from FDA since the turn of 2022. Humira, Skyrizi, and Rinvoq together accounted for nearly half of AbbVie’s top-line revenue in 2021 and the first half of 2022; and, given the rapid growth of the latter two, this state of affairs doesn’t look likely to change any time soon.
“We delivered another strong quarter with substantial progress for our new products and indications,” said AbbVie CEO Richard Gonzalez in the Q2 2022 earnings announcement. “Importantly, Skyrizi and Rinvoq continued their impressive ramps and are on pace to deliver approximately $7.5 billion in combined annual sales, underscoring their significant potential. The momentum of our business, combined with advances across our pipeline continue to support AbbVie’s promising long-term outlook.”
In its first full year with legacy Allergan sales, AbbVie’s top-line revenue in 2021 was $56.2 billion, a 22.7 percent improvement over the previous year. Net income more than doubled, from $4.62 billion to $11.55 billion, and earnings per share rose by $3.73 to $6.45. In first-half 2022, with the Allergan transaction off the lag side of the books, AbbVie’s top-line rose another 4.3 percent to $28.12 billion, with net income up 25.4 percent to $5.42 billion and EPS up 62 cents to $3.03.
Partnerships and acquisitions
In March, AbbVie completed the acquisition of Syndesi Therapeutics SA, which executives say will help to expand the company’s neuroscience portfolio. This acquisition gives AbbVie access to Syndesi’s portfolio of novel modulators of the synaptic vesicle protein 2A (SV2A), including its lead molecule SDI-118. The mechanism is currently being evaluated for the potential treatment of cognitive impairment and other symptoms associated with a range of neuropsychiatric and neurodegenerative disorders, such as Alzheimer’s disease and major depressive disorder. Under the terms of the agreement, AbbVie paid Syndesi shareholders a $130 million upfront payment with the potential for Syndesi shareholders to receive additional contingent payments of up to $870 million based on the achievement of certain predetermined milestones.
Also in March, AbbVie and Gedeon Richter Plc. announced a new joint development and license agreement to research, develop, and commercialize novel dopamine receptor modulators for the potential treatment of neuropsychiatric diseases. The collaboration is based on the results of preclinical research carried out by Richter and includes several new chemical entities selected for development. AbbVie and Richter have collaborated for 15 years on central nervous system (CNS) projects, including globally launched products such as cariprazine (marketed as Vraylar/Reagila).
Under the terms of the agreement, the collaboration includes both preclinical and clinical R&D activities with shared financing by the parties. Richter received an upfront cash payment, along with potential future development, regulatory and commercialization milestones. In addition, Richter may also receive sales-based royalties. AbbVie will have worldwide commercialization rights except for traditional markets of Richter, such as geographic Europe, Russia, other CIS countries, and Vietnam.
Also in March, AbbVie and Scripps Research, an independent, non-profit biomedical research and drug discovery institute, announced a global collaboration to develop potential novel, direct-acting antiviral treatments for COVID-19.
In May, AbbVie and Cugene Inc., a clinical-stage biotech company focused on developing next-generation precision immunology and oncology medicines to treat autoimmune disease and cancer, announced an exclusive worldwide license option agreement for CUG252, a potential best-in-class Treg-selective IL-2 mutein, as well as other novel IL-2 muteins, for the potential treatment of autoimmune and inflammatory diseases. Selective IL-2 muteins have the potential to represent a major advancement in the standard of care for patients with autoimmune and inflammatory diseases. Cugene’s lead candidate, CUG252, is an engineered IL-2 mutein designed to selectively activate and expand immune-suppressive Treg cells while reducing undesired IL-2 activity on other IL-2 receptor expressing cells for the treatment of autoimmune diseases. CUG252 is currently in a Phase I study in healthy volunteers.
Under the terms of the agreement, Cugene received an upfront payment of $48.5 million, and will also be eligible to receive development and regulatory milestones and a license option exercise payment if AbbVie exercises the option. In addition, Cugene may also receive commercialization and sales-based milestones and tiered royalties. AbbVie will receive an option to obtain an exclusive license for certain IL-2 muteins, including CUG252. During the option period, Cugene will conduct a Phase Ib study in patients with autoimmune/inflammatory disease. Upon exercise of the option, AbbVie will conduct all future clinical development, manufacturing and commercialization activities for CUG252.
AbbVie and iSTAR Medical SA in July announced a strategic deal to further develop and commercialize iSTAR’s MINIject device, a minimally invasive glaucoma surgical (MIGS) device for patients with glaucoma. This complementary alliance, leaders say, will support iSTAR Medical’s development and commercial efforts for MINIject, as well as provide an opportunity to expand AbbVie’s eye care business, building on its glaucoma portfolio, which includes drops, sustained release implants, and stent offerings.
Under the terms of the agreement, iSTAR Medical received a $60 million non-dilutive upfront payment and will continue to develop and commercialize MINIject until completion of the STAR-V clinical study. AbbVie will hold the exclusive right to acquire iSTAR Medical and lead subsequent global development and commercialization of the MINIject device. If AbbVie exercises the right to acquire iSTAR, the stockholders of iSTAR Medical would also be eligible to receive additional contingent payments of up to $475 million in a closing payment and upon achievement of certain predetermined milestones.
Product performance

It may not be the world’s best-selling pharmaceutical any more, but Humira still generated more than $20 billion in sales for AbbVie in 2021 and looks likely to do so again in 2022.
It may not be the world’s leading pharmaceutical product any more – the Pfizer/BioNTech COVID vaccine ended that run – but the autoimmune drug Humira still generated $20.69 billion in sales for AbbVie in 2021, an improvement of 4.3 percent compared with 2020. According to company leaders, this was primarily driven by market growth across therapeutic categories, partially offset by direct biosimilar competition in certain international markets. In the United States, Humira sales increased 8 percent in 2021 driven by market growth across all indications. This increase was partially offset by slightly lower market share following corresponding market share gains of AbbVie stable-mates Skyrizi and Rinvoq. Internationally, Humira revenue decreased 13 percent in 2021 primarily driven by direct biosimilar competition in certain international markets. In the first half of 2022, Humira sales edged up another 1.7 percent to $10.1 billion.
The oncologic Imbruvica generated $5.41 billion in sales for AbbVie in 2021, up 1.8 percent from 2020. AbbVie executives say this was as a result of modest favorable pricing in the United States and increased collaboration revenue, partially offset by lower new patient starts due to the COVID-19 pandemic and share loss in the United States. In the first half of 2022, Imbruvica sales declined by 12.5 percent to $2.32 billion, as a result of decreased market demand and lower new patient starts in the United States.
In August, FDA approved the use of Imbruvica for the treatment of pediatric patients one year and older with chronic graft versus host disease (cGVHD) after failure of one or more lines of systemic therapy. The approval was AbbVie’s first pediatric indication for Imbruvica, marking the 12th FDA approval for Imbruvica and the first Bruton’s tyrosine kinase inhibitor (BTKi) treatment approved for a pediatric patient population. Imbruvica is now the first approved treatment option for children under 12 years of age suffering from cGVHD. The approval was primarily based on positive results from the iMAGINE Phase I/II clinical trial. The iMAGINE study demonstrated an Overall Response Rate through week 25 of 60 percent in patients median age 13 years (range, 1-19 years) with relapsed/refractory (R/R) moderate to severe cGVHD.5 The median duration of response was 5.3 months.
Sales of the autoimmune drug Skyrizi nearly doubled in 2021, from $1.59 billion to $2.94 billion. According to company leaders, this was primarily driven by continued strong volume and market share uptake since launch in 2019 as a treatment for plaque psoriasis as well as market growth over the prior year. In the first half of 2022, Skyrizi sales were up another 75.6 percent to $2.19 billion.
In January, FDA approved Skyrizi for the treatment of adults with active psoriatic arthritis. The FDA approval was supported by data from two pivotal studies, KEEPsAKE-1 and KEEPsAKE-2, which evaluated the efficacy and safety of Skyrizi in adults with active PsA, including those who had responded inadequately or were intolerant to biologic therapy and/or non-biologic disease-modifying antirheumatic drugs (DMARDs). Across the two Phase III studies, Skyrizi met the primary endpoint of ACR20 response at week 24 compared to placebo and demonstrated significant improvements across several other manifestations of PsA, including swollen, tender, and painful joints.
In June, FDA approved Skyrizi as the first and only specific interleukin-23 (IL-23) inhibitor for the treatment of adults with moderately to severely active Crohn’s disease (CD). In two induction and one maintenance clinical trials, Skyrizi demonstrated significant improvements in endoscopic response (defined as a decrease of greater than 50 percent from the baseline Simple Endoscopic Score in CD [SES-CD] or for patients with isolated ileal disease and SES-CD of 4, at least a 2-point reduction from baseline) and clinical remission (defined as a Crohn’s Disease Activity Index [CDAI] of less than 150), compared to placebo, as both an induction and maintenance therapy.

Sales of Skyrizi nearly doubled in 2021 to $2.94 billion; the autoimmune product has earned new approvals from FDA in psoriatic arthritis and Crohn’s during 2022.
The co-primary endpoints of the clinical trials were endoscopic response and clinical remission. In the 12-week induction studies, ADVANCE and MOTIVATE, a significantly greater proportion of patients treated with Skyrizi achieved endoscopic response and clinical remission compared to placebo. As early as week 4, clinical response (defined as a 100-point reduction in CDAI) and clinical remission were achieved in a significantly greater proportion of patients receiving Skyrizi as compared to placebo. In the 52-week maintenance trial, FORTIFY, a significantly greater proportion of patients achieved the co-primary endpoints of endoscopic response and clinical remission as compared with the placebo group (risankizumab induction responders) after one year.
Both sides of AbbVie’s Botox portfolio, the Cosmetic and the Therapeutic, enjoyed impressive growth in 2021, with Cosmetic sales more than doubling from $1.11 billion to $2.23 billion and Therapeutic up 76.7 percent to $2.45 billion. According to AbbVie leaders, this was due to increased brand investment and strong recovery from the COVID-19 pandemic. Sales were also favorably impacted by a full period of Allergan results in 2021 compared to the prior year. In the first half of 2022, Botox Cosmetic sales rose another 25.9 percent to $1.34 billion and Botox Therapeutic sales were up 13.8 percent to $1.29 billion.
The leukemia/lymphoma treatment Venclexta enjoyed a sales bounce of 36.1 percent in 2021, to $1.82 billion. According to company leaders, this was primarily due to continued expansion of Venclexta for the treatment of patients with first-line CLL, relapsed/refractory CLL, and first-line AML. In the first half of 2022, Venclexta sales were up another 16.4 percent to $978 billion.
In June, AbbVie announced five-year follow-up results from the Phase III CLL14 trial, finding that more than 60 percent of patients with previously untreated chronic lymphocytic leukemia (CLL) who had received one-year fixed-duration combination treatment of Venclyxto/Venclexta plus obinutuzumab (Gazyva) continued to show longer progression-free survival (PFS) and higher rates of undetectable minimal residual disease (MRD) after four years off treatment.
Data shows that after more than five years of median follow-up (65.4 months), PFS remained significantly superior among patients treated with the Venclyxto/Venclexta and obinutuzumab combination compared to the chlorambucil and obinutuzumab chemotherapy regimen (n=432; median NR versus 36.4 months). The therapies were administered for a fixed-duration of 12 months for Venclyxto/Venclexta in combination with six cycles of obinutuzumab. At five years after randomization, the estimated PFS rate after one-year fixed-duration treatment was 62.6 percent for the Venclyxto/Venclexta-based combination compared to 27.0 percent for the chlorambucil combination. The improvement in PFS was maintained across all risk groups, including patients with TP53 mutation/deletion and unmutated IGHV status.
Vraylar, indicated for the treatment of schizophrenia, bipolar I disorder, and bipolar depression, generated $1.73 billion in sales for AbbVie in 2021, nearly double its $951 million from the year before. According to AbbVie executives, this was due to higher market share and market growth. Sales were also favorably impacted by a full period of Allergan results in 2021 compared to the prior year. In the first half of 2022, sales of Vraylar rose another 18.1 percent to $919 million.
In February, AbbVie submitted a supplemental New Drug Application for Vraylar to FDA for the adjunctive treatment of major depressive disorder (MDD) in patients who are receiving ongoing antidepressant therapy. Supporting this submission, a Phase III study 3111-301-001 showed a clinically and statistically significant change from baseline to week six in the Montgomery-Åsberg Depression Rating Scale (MADRS) total score for patients treated with Vraylar at 1.5 mg/day compared with placebo. A second registration-enabling study, RGH-MD-75, showed a clinically and statistically significant change from baseline to week eight in the MADRS total score for patients treated with Vraylar at 2-4.5 mg/day compared with placebo.

Rinvoq sales more than doubled to $1.65 billion in 2021; the drug earned new approvals in atopic dermatitis, ulcerative colitis, and ankylosing spondylitis from FDA in 2022.
The autoimmune product Rinvoq more than doubled its sales in 2021, jumping from $731 million to $1.65 billion. AbbVie leaders said this was primarily driven by continued strong volume and market share uptake since launch in 2019 for the treatment of moderate to severe rheumatoid arthritis as well as market growth over the prior year. Sales were also favorably impacted by recent regulatory approvals and expansion of Rinvoq for the treatment of psoriatic arthritis, atopic dermatitis and ankylosing spondylitis in certain international markets. First-half 2022 Rinvoq sales rose another 55.2 percent to $1.06 billion.
In January, FDA approved Rinvoq for the treatment of moderate-to-severe atopic dermatitis in adults and children 12 years of age and older whose disease did not respond to previous treatment and is not well controlled with other pills or injections, including biologic medicines, or when use of other pills or injections is not recommended. The approval was supported by efficacy and safety data from one of the largest registrational Phase III programs for atopic dermatitis with more than 2,500 patients evaluated across three studies. About 52 percent of the patients had prior exposure to systemic atopic dermatitis treatment. These studies evaluated the efficacy and safety of Rinvoq monotherapy (Measure Up 1 and 2) and with topical corticosteroids (AD Up), compared to placebo, in adults and children 12 years of age and older with moderate to severe atopic dermatitis. Across the three atopic dermatitis pivotal studies, Rinvoq (15 mg and 30 mg, once daily) monotherapy and with topical corticosteroids met all primary and secondary endpoints at week 16, with some patients achieving higher levels of skin clearance (EASI 90 and 100).
In February, AbbVie announced positive top-line results from U-EXCEL, a Phase III induction study, showing Rinvoq (45 mg once daily) achieved both primary endpoints of clinical remission and endoscopic response at week 12. A significantly greater proportion of patients treated with a 12-week induction regimen of Rinvoq achieved clinical remission per CDAI at week 12 compared to placebo (49 percent versus 29 percent). Similar results were observed with clinical remission per SF/AP (51 percent in Rinvoq-treated patients versus 22 percent in placebo-treated patients). At week 12, a significantly greater proportion of patients treated with upadacitinib 45 mg achieved endoscopic response compared to the placebo group (46 percent versus 13 percent).
In March, FDA approved Rinvoq for the treatment of adults with moderately to severely active ulcerative colitis (UC) who have had an inadequate response or intolerance to one or more tumor necrosis factor (TNF) blockers. This FDA approval was the first indication for Rinvoq in gastroenterology and was supported by efficacy and safety data from three Phase III randomized, double-blind, placebo-controlled clinical studies.
The two induction studies (U-ACHIEVE and U-ACCOMPLISH) utilized Rinvoq 45 mg once daily for 8 weeks, and then 15 mg or 30 mg once daily for the maintenance study (U-ACHIEVE maintenance) through 52 weeks. Across all clinical trials, significantly more patients treated with Rinvoq achieved clinical remission at weeks 8 and 52, the primary endpoint based on the mMS: stool frequency subscore (SFS) ≤ 1 and not greater than Baseline, rectal bleeding subscore (RBS) = 0, endoscopy subscore (ES) of ≤ 1 without friability, compared to placebo. In addition, the studies met all ranked secondary endpoints, including endoscopic improvement and histologic-endoscopic mucosal improvement (HEMI), as well as corticosteroid-free clinical remission in the maintenance study.
In April, FDA approved Rinvoq for the treatment of adults with active ankylosing spondylitis (AS) who have had an inadequate response or intolerance to one or more tumor necrosis factor (TNF) blockers. The approval was supported by efficacy and safety data from the Phase III SELECT-AXIS 2 clinical trial (Study 1) evaluating Rinvoq in patients who had an inadequate response or intolerance to one or two biologic disease-modifying anti-rheumatic drugs (bDMARDs) and the Phase II/III SELECT-AXIS 1 clinical trial evaluating Rinvoq in patients who were naïve to bDMARDs and had an inadequate response or intolerance to at least two nonsteroidal anti-inflammatory drugs (NSAIDs). In both the SELECT-AXIS 1 and SELECT-AXIS 2 clinical trials, a significantly greater proportion of patients receiving Rinvoq 15 mg achieved an ASAS40 response, the primary endpoint, (51 percent and 44.5 percent, respectively) compared to those receiving placebo (26 percent and 18.2 percent, respectively) at week 14. Clinical responses were observed as early as week four in SELECT-AXIS 2 for ASAS40.
In May, AbbVie announced positive topline results from U-ENDURE, its Phase III maintenance study evaluating Rinvoq in adult patients with moderate-to-severe Crohn’s disease who had an inadequate response or were intolerant to a conventional or biologic therapy. The results showed that more patients treated with either dose of Rinvoq (15 mg or 30 mg once daily) achieved the co-primary endpoints of endoscopic response and clinical remission, as well as the secondary endpoint of endoscopic remission, at one year (week 52) compared to placebo. Results from the U-ENDURE maintenance study, in addition to results from the U-EXCEED and U-EXCEL induction studies, will be included in future regulatory submissions.
A significantly higher proportion of patients who received Rinvoq 15 mg or 30 mg achieved clinical remission per the CDAI at week 52: 37 and 48 percent, respectively, versus 15 percent in the placebo group. Results also showed that 36 and 46 percent of patients who received Rinvoq 15 mg and 30 mg, respectively, achieved clinical remission at week 52 per SF/AP compared to 14 percent in the placebo group. At week 52, 28 and 40 percent of patients who received Rinvoq 15 mg and 30 mg, respectively, achieved endoscopic response compared to 7 percent of patients who received placebo. In addition, 19 and 29 percent of patients who received Rinvoq 15 mg and 30 mg achieved endoscopic remission, respectively, compared to 5 percent of patients in the placebo group. A significantly higher proportion of patients who received Rinvoq 15 mg or 30 mg achieved corticosteroid-free clinical remission per CDAI and per SF/AP compared to placebo at week 52 among patients taking corticosteroids at baseline.
In July, the European Commission approved Rinvoq for the treatment of active non-radiographic axial spondyloarthritis (nr-axSpA) in adult patients with objective signs of inflammation, as indicated by elevated C-reactive protein (CRP) and/or magnetic resonance imaging (MRI), who have responded inadequately to nonsteroidal anti-inflammatory drugs (NSAIDs). The approval was based on results from the Phase III SELECT-AXIS 2 nr-axSpA clinical trial. Study results showed a significantly greater proportion of patients receiving Rinvoq achieved an Assessment of SpondyloArthritis international Society 40 percent (ASAS40) response at week 14 (45 percent versus 23 percent) compared to placebo. Statistical significance was also achieved in 12 of the 14 multiplicity-controlled secondary endpoints compared to placebo at week 14.
Also in July, the European Commission approved Rinvoq for the treatment of adult patients with moderately to severely active ulcerative colitis who have had an inadequate response, lost response or were intolerant to either conventional therapy or a biologic agent. The approval was supported by data from two induction studies, U-ACHIEVE induction and U-ACCOMPLISH, and one maintenance study, U-ACHIEVE maintenance. Statistical significance was achieved for the primary endpoint and all secondary endpoints with Rinvoq 45 mg in the two induction studies and both Rinvoq 15 mg and 30 mg in the maintenance study.
Sales of the migraine treatment Ubrelvy more than quadrupled in 2021, from $125 million to $552 million. Company leaders said this was primarily due to increased volume and market share uptake since launch in 2020. In first-half 2022, Ubrelvy sales rose another 56 percent to $323 million.
In the pipeline
In January, FDA granted Breakthrough Therapy Designation to investigational telisotuzumab vedotin (Teliso-V) for the treatment of patients with advanced/metastatic epidermal growth factor receptor (EGFR) wild type, nonsquamous non-small cell lung cancer (NSCLC) with high levels of c-Met overexpression whose disease has progressed on or after platinum-based therapy.
This BTD designation was supported by data from LUMINOSITY (Study M14-239), an ongoing Phase II study designed to identify the target NSCLC populations that overexpress c-Met best suited for Teliso-V monotherapy in the second line or third line setting, and then to expand the groups to further evaluate efficacy in the selected populations. The primary endpoint is overall response rate (ORR) per central review in patients with ≥ 12 weeks follow-up. Among patients with EGFR WT nonsquamous NSCLC, ORR was 53.8 percent in the c-Met high group and 25.0 percent in the c-Met intermediate group at a previously reported interim analysis. Teliso-V is also being evaluated in combination with osimertinib in the ongoing Phase I study M14-237 in patients with previously treated c-Met overexpressing NSCLC. In addition, it will be further evaluated as monotherapy in patients with previously treated c-Met overexpressing NSCLC in the randomized Phase III study TeliMET NSCLC-01 (Study M18-868).
In April, the AbbVie company Allergan announced that the Phase III VIRGO trial evaluating the safety and efficacy of investigational twice-daily administration of Vuity 1.25% in adults with presbyopia met its primary efficacy endpoint, improving near vision without compromising distance vision at Hour 9 (3 hours after the second drop) on Day 14. Approved by FDA in October 2021 for once-daily use, Vuity is the first and only eye drop to treat age-related blurry near vision in adults.
In the VIRGO Phase III clinical trial, a total of 230 participants aged 40 to 55 years old with presbyopia were randomized in a one-to-one ratio of vehicle (placebo) to Vuity, receiving two drops in each eye per day for 14 days, with the second drop at Hour 6 (6 hours after the first drop). The study met its primary endpoint, demonstrating a statistically significant proportion of participants treated with Vuity twice daily gained three lines (the ability to read three additional lines on a near vision chart) or more in mesopic (low light), high contrast, binocular Distance Corrected Near Visual Acuity (DCNVA) with no more than 5-letter loss in low light Corrected Distance Visual Acuity (CDVA) at Day 14, Hour 9 (3 hours after the second drop) compared to the vehicle (placebo).
In May, AbbVie submitted a New Drug Application to FDA for ABBV-951 (foscarbidopa/foslevodopa) for the treatment of motor fluctuations in patients with advanced Parkinson’s disease. The submission was based on results from a Phase III, head-to-head, randomized and controlled clinical trial demonstrating statistically significant improvement in “on” time without troublesome dyskinesia compared to oral immediate-release carbidopa/levodopa (CD/LD).
In June, AbbVie announced primary results from the large B-cell lymphoma (LBCL) expansion cohort in the EPCORE NHL-1 phase II clinical trial evaluating epcoritamab. In this study, epcoritamab demonstrated efficacy with durable responses in patients who had previously received at least two prior lines of anti-lymphoma therapy including chimeric antigen receptor (CAR) T-cell therapy.
The study cohort, which included 157 relapsed/refractory LBCL patients, previously treated with a median of three lines of prior therapy, demonstrated an overall response rate of 63 percent and a complete response rate of 39 percent. Baseline characteristics included 61 percent of patients who were refractory to primary treatment, 20 percent who had prior autologous stem cell transplantation (ASCT), and 39 percent who were treated with CAR T-cell therapy (75 percent of those refractory to CAR T). Patients enrolled in the study who were naïve to CAR T-cell therapy achieved a 69 percent ORR and a 42 percent CR and patients who received prior CAR T-cell therapy achieved a 54 percent ORR and a 34 percent CR. After a median follow up of 10.7 months, the median duration of response (mDOR) was estimated to be 12 months, while the mDOR among patients achieving a CR was not reached, with 89 percent still in CR at nine months.
Also in June, AbbVie announced new data from Cohort 3 of its Phase II REFINE study of investigational navitoclax in combination with ruxolitinib in JAK inhibitor naïve patients with myelofibrosis (MF), a rare and difficult-to-treat blood cancer. These preliminary findings show spleen volume and symptomatic improvement in this cohort. The primary endpoint was spleen volume reduction of ≥35 percent (SVR35) from baseline at week 24.2 Key secondary endpoints include ≥50 percent reduction in total symptom score (TSS50) at week 24, anemia response and BMF reduction.
In the results, SVR35 was achieved by 63 percent of evaluable patients at week 24 (20/32) and by 78 percent at any time on treatment (25/32). At week 24, 41 percent (11/27) of evaluable patients with measurable baseline symptoms reached TSS50; 67 percent of patients (18/27) met this endpoint at any time during the study. In this cohort, 35 percent of evaluable patients, with available fibrosis grade at baseline and during the study, (9/26) achieved reduction in BMF by ≥1 grade at any time during the study with three patients experiencing ≥2 grade reductions in BMF. Additionally, 40 percent of patients evaluable for anemia response (6/15) experienced improvement in anemia, a common clinical feature of MF.
Also in June, AbbVie submitted a supplemental New Drug Application for Qulipta to FDA to support the preventive treatment of chronic migraine in adults. If approved, Qulipta would be the first gepant (oral calcitonin gene-related peptide [CGRP] receptor antagonist) approved for the broad indication of the preventive treatment of migraine, including episodic and chronic. The sNDA submission includes data from the pivotal Phase III PROGRESS trial in patients with chronic migraine, which supplements the existing data in episodic migraine.
The Phase III PROGRESS trial met its primary endpoint of statistically significant reduction from baseline in mean monthly migraine days compared to placebo across the 12-week treatment period in adults with chronic migraine. The trial also demonstrated that treatment with Qulipta 60 mg once daily (QD) and 30 mg daily (BID) resulted in statistically significant improvements in all six secondary endpoints. This includes a key secondary endpoint that measured the proportion of patients that achieved at least a 50 percent reduction in mean monthly migraine days across the 12-week treatment period.
In July, AbbVie submitted a marketing authorization application to the European Medicines Agency for Qulipta for the prophylaxis of migraine in adult patients who have at least four migraine days per month. The application was supported by the pivotal Phase III ADVANCE and PROGRESS studies evaluating the safety, efficacy, and tolerability of Qulipta in adult patients with episodic migraine and chronic migraine, respectively.
The Phase III multicenter, randomized, double-blind, placebo-controlled, parallel-group ADVANCE trial evaluated the efficacy, safety, and tolerability of once daily (QD) oral Qulipta for the prophylaxis of episodic migraine. The study met its primary endpoint of a statistically significant reduction in mean monthly migraine days across the 12-week treatment period compared to placebo. This was found across all active treatment arms of Qulipta – 10 mg, 30 mg, and 60 mg QD doses. The adult patients enrolled met the International Classification of Headache Disorders (ICHD) criteria for a diagnosis of migraine with or without aura. The study also found that a greater proportion of Qulipta-treated participants achieved at least a 50 percent reduction in mean monthly migraine days for all doses compared to placebo and met other key secondary endpoints.
depression pandemic covid-19 vaccine treatment fda clinical trials preclinical therapy recovery european europe russiaGovernment
Stock Market Today: Stocks turn lower as factory inflation spikes, retail sales miss target
Stocks will navigate the last major data releases prior to next week’s Fed rate meeting in Washington.
Share this:


Check back for updates throughout the trading day
U.S. stocks edged lower Thursday following a trio of key economic releases that have added to the current inflation puzzle as investors shift focus to the Federal Reserve's March policy meeting next week in Washington.
Updated at 9:59 AM EDT
Red start
Stocks are now falling sharply following the PPI inflation data and retail sales miss, with the S&P 500 marked 18 points lower, or 0.36%, in the opening half hour of trading.
The Dow, meanwhile, was marked 92 points lower while the Nasdaq slipped 67 points.
Treasury yields are also on the move, with 2-year notes rising 5 basis points on the session to 4.679% and 10-year notes pegged 7 basis points higher at 4.271%.
The probability of a June rate cut has moved below 60% after the higher-than-expected CPI/PPI reports. A week ago this probability was 74% and a month ago it was 82%. pic.twitter.com/9W01oWU96G
— Charlie Bilello (@charliebilello) March 14, 2024
Updated at 9:44 AM EDT
Under Water
Under Armour (UAA) shares slumped firmly lower in early trading following the sportswear group's decision to bring back founder Kevin Plank as CEO, replacing the outgoing Stephanie Linnartz.
Plank, who founded Under Armour in 1996, left the group in May of 2021 just weeks before the group revealed that it was co-operating with investigations from both the Securities and Exchange Commission and the U.S. Department of Justice into the company's revenue recognition accounting.
Under Armour shares were marked 10.6% lower in early trading to change hands at $7.21 each.
Updated at 9:22 AM EDT
Steely resolve
U.S. Steel (X) shares extended their two-day decline Thursday, falling 5.75% in pre-market trading following multiple reports that suggest President Joe Biden will push to prevent Japan's Nippon Steel from buying the Pittsburgh-based group.
Both Reuters and the Associated Press have said Biden will express his views to Prime Minister Kishida Yuko ahead of a planned State Visit next month at the White House.
Related: US Steel soars on $15 billion Nippon Steel takeover; United Steelworkers slams deal
Updated at 8:52 AM EDT
Clear as mud
Retail sales rebounded last month, but the overall tally of $700.7 billion missed Street forecasts and suggests the recent uptick in inflation could be holding back discretionary spending.
A separate reading of factory inflation, meanwhile, showed prices spiking by 1.6%, on the year, and 0.6% on the month, amid a jump in goods prices.
U.S. stocks held earlier gains following the data release, with futures tied to the S&P 500 indicating an opening bell gain of 10 points, while the Dow was called 140 points higher. The Nasdaq, meanwhile, is looking at a more modest 40 point gain.
Benchmark 10-year Treasury note yields edged 3 basis points lower to 4.213% while two-year notes were little-changed at 4.626%.
The #PPI troughed 8 months ago, yet the economic consensus and even the #Fed believes #inflation has been conquered. Forget the forecasts for multiple rate cuts. pic.twitter.com/ZNIiKLWdFA
— Richard Bernstein Advisors (@RBAdvisors) March 14, 2024
Stock Market Today
Stocks finished lower last night, with the S&P 500 ending modestly in the red and the Nasdaq falling around 0.5%. The declines came amid an uptick in Treasury yields tied to concern that inflation pressures have failed to ease over the opening months of the year.
A better-than-expected auction of $22 billion in 30-year bonds, drawing the strongest overall demand since last June, steadied the overall market, but stocks still slipped into the close with an eye towards today's dataset.
The Commerce Department will publish its February reading of factory-gate inflation at 8:30 am Eastern Time. Analysts are expecting a slowdown in the key core reading, which feeds into the Fed's favored PCE price index.
Retail sales figures for the month are also set for an 8:30 am release as investors search for clues on consumer strength, tied to a resilient job market. Those factors could give the Fed more justification to wait until the summer months to begin the first of its three projected rate cuts.
"The case for a gradual but sustained slowdown in growth in consumers’ spending from 2023’s robust pace is persuasive," said Ian Shepherdson of Pantheon Macroeconomics.
"Most households have run down the excess savings accumulated during the pandemic, while the cost of credit has jumped and last year’s plunge in home sales has depressed demand housing-related retail items like furniture and appliances," he added.
Benchmark 10-year Treasury yields are holding steady at 4.196% heading into the start of the New York trading session, while 2-year notes were pegged at 4.628%.
With Fed officials in a quiet period, requiring no public comments ahead of next week's meeting in Washington, the U.S. dollar index is trading in a narrow range against its global peers and was last marked 0.06% higher at 102.852.
On Wall Street, futures tied to the S&P 500 are indicating an opening bell gain of around 19 points, with the Dow Jones Industrial Average indicating a 140-point advance.
The tech-focused Nasdaq, which is up 7.77% for the year, is priced for a gain of around 95 points, with Tesla (TSLA) once again sliding into the red after ending the Wednesday session at a 10-month low.
In Europe, the regionwide Stoxx 600 was marked 0.35% higher in early Frankfurt trading, while Britain's FTSE 100 slipped 0.09% in London.
Overnight in Asia, the Nikkei 225 gained 0.29% as investors looked to a key series of wage negotiation figures from key unions that are likely to see the biggest year-on-year pay increases in three decades.
The broader MSCI ex-Japan benchmark, meanwhile, rose 0.18% into the close of trading.
Related: Veteran fund manager picks favorite stocks for 2024
bonds pandemic dow jones sp 500 nasdaq ftse stocks rate cut fed federal reserve home sales white house japan europeGovernment
Trump nearly derailed democracy once − here’s what to watch out for in reelection campaign
Donald Trump tried to overturn the 2020 election results. But the work of others, from lawmakers to judges to regular citizens, stopped him. There are…
Share this:


Elections are the bedrock of democracy, essential for choosing representatives and holding them accountable.
The U.S. is a flawed democracy. The Electoral College and the Senate make voters in less populous states far more influential than those in the more populous: Wyoming residents have almost four times the voting power of Californians.
Ever since the Civil War, however, reforms have sought to remedy other flaws, ensuring that citizenship’s full benefits, including the right to vote, were provided to formerly enslaved people, women and Native Americans; establishing the constitutional standard of one person, one vote; and eliminating barriers to voting through the 1965 Voting Rights Act.
But the Supreme Court has, in recent years, narrowly construed the Voting Rights Act and limited courts’ ability to redress gerrymandering, the drawing of voting districts to ensure one party wins.
The 2020 election revealed even more disturbing threats to democracy. As I explain in my book, “How Autocrats Seek Power,” Donald Trump lost his reelection bid in 2020 but refused to accept the results. He tried every trick in the book – and then some – to alter the outcome of this bedrock exercise in democracy.
A recent New York Times story reports that when it comes to Trump’s time in office and his attempt to overturn the 2020 election, “voters often have a hazy recall of one of the most tumultuous periods in modern politics.” This, then, is a refresher about Trump’s handling of the election, both before and after Nov. 3, 2020.
Trump began with a classic autocrat’s strategy – casting doubt on elections in advance to lay the groundwork for challenging an unfavorable outcome.
Despite his efforts, Trump was unable to control or change the election results. And that was because of the work of others to stop him.
Here are four things Trump tried to do to flip the election in his favor – and examples of how he was stopped, both by individuals and democratic institutions.
Anticipating defeat
Expecting to lose in November 2020, in part because of his disastrous handling of the Covid-19 pandemic, Trump proclaimed that “all over the country, especially in California, voter fraud is rampant.” He called mail ballots “a very dangerous thing.” Jared Kushner, his son-in-law and aide, declined to “commit one way or the other” about whether the election would be held in November, because of the COVID pandemic. No efforts to postpone the election ensued.
Trump warned that Russia and China would “be able to forge ballots,” a myth echoed by Attorney General William Barr. Trump illegally threatened to have law enforcement officers at polling places. He falsely asserted that Kamala Harris “doesn’t meet the requirements” for serving as vice president because her parents were immigrants. Asked if he would agree to a transition if he lost, he responded: “There won’t be a transfer, frankly. There’ll be a continuation.”
Threatening litigation
Aware that polls showed Biden ahead by 8 percentage points, Trump declared, “As soon as that election is over, we’re going in with our lawyers,” and they did just that. Adviser Steve Bannon correctly predicted that on Election Night, “Trump’s gonna walk into the Oval (Office), tweet out, ‘I’m the winner. Game over, suck on that.’”
Trump followed the script, asserting at 2:30 am: “we did win this election. … This is a major fraud in our nation,” though the actual results weren’t clear until days later, when, on Nov. 7, the networks declared Biden had won.
Although many advisers said he had lost, Trump kept claiming fraud, repeating Rudy Giuliani’s false allegation that Dominion election machines had switched votes – a lie for which Fox News agreed to pay $787 million to settle the defamation case brought by Dominion.
Taking direct action
Trump allies pressured state legislators to create false, “alternative” slates of electors as a key strategy for overturning the election. Trump contemplated declaring an emergency, ordering the military to seize voting machines and replacing the attorney general with a yes-man who would pressure state legislatures to change their electoral votes.
Encouraging violence
Trump summoned supporters to protest the Jan. 6 certification by Congress, boasted it would be “wild,” and encouraged them to march on the Capitol and “fight like hell,” promising to accompany them. Once they had attacked the Capitol, he delayed for four hours before asking them to stop.
Yet Trump’s efforts to overturn the election failed.

Resisting Trump
Trump claimed that voting by mail produced rampant fraud, but state legislatures let voters vote by mail or in drop boxes because of the pandemic. Postal Service workers delivered those ballots despite actions taken by Trump’s postmaster general, Louis DeJoy, that made processing and delivery more difficult.
DeJoy denied any sabotage in testimony before Congress.
Most state election officials, regardless of party, loyally did their jobs, resisting Trump’s pressure to falsify the outcome. Courts rejected all but one of Trump’s 62 lawsuits aimed at overturning the election. Government lawyers refused to invoke the Insurrection Act and authorize the military to seize voting machines. The military remained scrupulously apolitical. And Vice President Mike Pence presided over the certification, in which 43 Republican senators and 75 Republican representatives joined all the Democrats to declare Biden the winner.
That experience contains invaluable lessons about what to expect in 2024 and how to defend the integrity of elections.
Richard L. Abel does not work for, consult, own shares in or receive funding from any company or organization that would benefit from this article, and has disclosed no relevant affiliations beyond their academic appointment.
white house congress senate trump pence pandemic covid-19 russia chinaGovernment
For-profit nursing homes are cutting corners on safety and draining resources with financial shenanigans − especially at midsize chains that dodge public scrutiny
Owners of midsize nursing home chains drain billions from facilities, hiding behind opaque accounting practices and harming the elderly as government,…
Share this:

{kind=link}
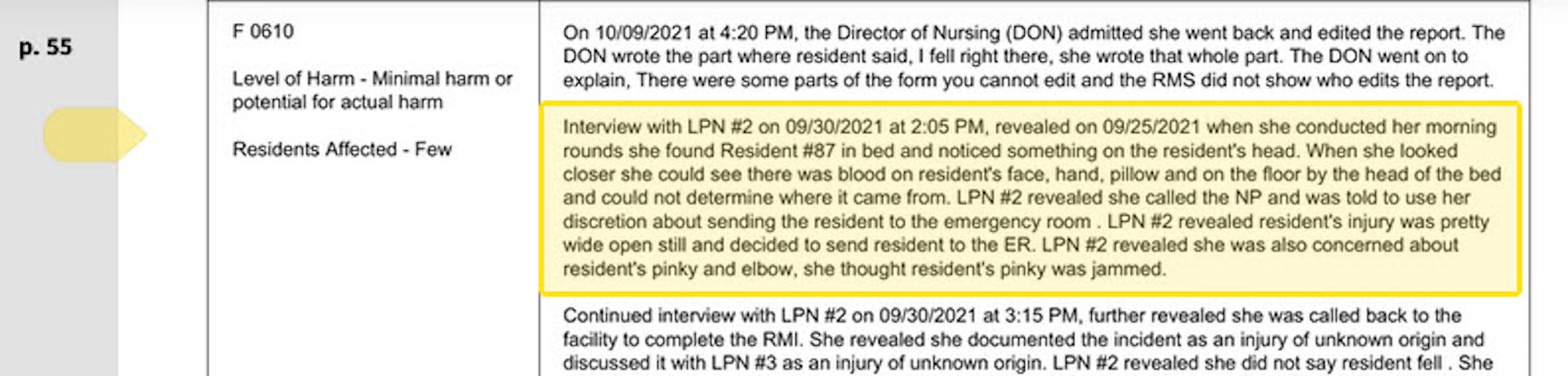
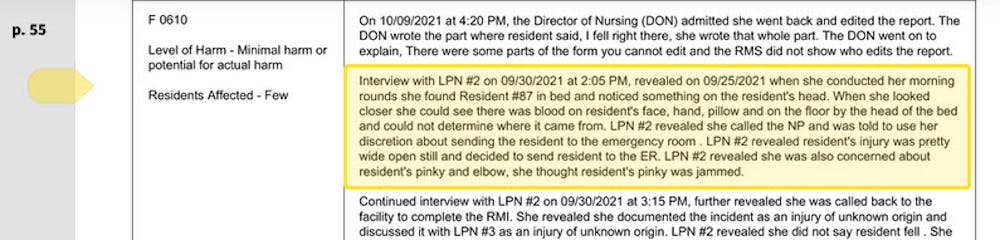
The care at Landmark of Louisville Rehabilitation and Nursing was abysmal when state inspectors filed their survey report of the Kentucky facility on July 3, 2021.
Residents wandered the halls in a facility that can house up to 250 people, yelling at each other and stealing blankets. One resident beat a roommate with a stick, causing bruising and skin tears. Another was found in bed with a broken finger and a bloody forehead gash. That person was allowed to roam and enter the beds of other residents. In another case, there was sexual touching in the dayroom between residents, according to the report.
Meals were served from filthy meal carts on plastic foam trays, and residents struggled to cut their food with dull plastic cutlery. Broken tiles lined showers, and a mysterious black gunk marred the floors. The director of housekeeping reported that the dining room was unsanitary. Overall, there was a critical lack of training, staff and supervision.
The inspectors tagged Landmark as deficient in 29 areas, including six that put residents in immediate jeopardy of serious harm and three where actual harm was found. The issues were so severe that the government slapped Landmark with a fine of over $319,000 − more than 29 times the average for a nursing home in 2021 − and suspended payments to the home from federal Medicaid and Medicare funds.
{kind=link}
Persistent problems
But problems persisted. Five months later, inspectors levied six additional deficiencies of immediate jeopardy − the highest level − including more sexual abuse among residents and a certified nursing assistant pushing someone down, bruising the person’s back and hip.
Landmark is just one of the 58 facilities run by parent company Infinity Healthcare Management across five states. The government issued penalties to the company almost 4½ times the national average, according to bimonthly data that the Centers for Medicare & Medicaid Services first started to make available in late 2022. All told, Infinity paid nearly $10 million in fines since 2021, the highest among nursing home chains with fewer than 100 facilities.
Infinity Healthcare Management and its executives did not respond to multiple requests for comment.
Such sanctions are nothing new for Infinity or other for-profit nursing home chains that have dominated an industry long known for cutting corners in pursuit of profits for private owners. But this race to the bottom to extract profits is accelerating despite demands by government officials, health care experts and advocacy groups to protect the nation’s most vulnerable citizens.
To uncover the reasons why, The Conversation’s investigative unit Inquiry delved into the nursing home industry, where for-profit facilities make up more than 72% of the nation’s nearly 14,900 facilities. The probe, which paired an academic expert with an investigative reporter, used the most recent government data on ownership, facility information and penalties, combined with CMS data on affiliated entities for nursing homes.
The investigation revealed an industry that places a premium on cost cutting and big profits, with low staffing and poor quality, often to the detriment of patient well-being. Operating under weak and poorly enforced regulations with financially insignificant penalties, the for-profit sector fosters an environment where corners are frequently cut, compromising the quality of care and endangering patient health. Meanwhile, owners make the facilities look less profitable by siphoning money from the homes through byzantine networks of interconnected corporations. Federal regulators have neglected the problem as each year likely billions of dollars are funneled out of nursing homes through related parties and into owners’ pockets.
More trouble at midsize
Analyzing newly released government data, our investigation found that these problems are most pronounced in nursing homes like Infinity − midsize chains that operate between 11 and 100 facilities. This subsection of the industry has higher average fines per home, lower overall quality ratings, and are more likely to be tagged with resident abuse compared with both the larger and smaller networks. Indeed, while such chains account for about 39% of all facilities, they operate 11 of the 15 most-fined facilities.
With few impediments, private investors who own the midsize chains have quietly swooped in to purchase underperforming homes, expanding their holdings even further as larger chains divest and close facilities. As a result of the industry’s churn of facility ownership, over one fifth of the country’s nursing facilities changed ownership between 2016 and 2021, four times more changes than hospitals.
A 2023 report by Good Jobs First, a nonprofit watchdog, noted that a dozen of these chains in the midsize range have doubled or tripled in size while racking up fines averaging over $100,000 per facility since 2018. But unlike the large, multistate chains with easily recognizable names, the midsize networks slip through without the same level of public scrutiny, The Conversation’s investigations unit found.
“They are really bad, but the names − we don’t know these names,” said Toby Edelman, senior policy attorney with the Center for Medicare Advocacy, a nonprofit law organization.
“When we used to have those multistate chains, the facilities all had the same name, so you know what the quality is you’re getting,” she said. “It’s not that good − but at least you know what you’re getting.”
In response to The Conversation’s findings on nursing homes and request for an interview, a CMS spokesperson emailed a statement that said the CMS is “unwavering in its commitment to improve safety and quality of care for the more than 1.2 million residents receiving care in Medicare- and Medicaid-certified nursing homes.”
The statement pointed to data released by the oversight body on mergers, acquisitions, consolidations and changes of ownership in April 2023 along with additional ownership data released the following September. CMS also proposed a rule change that aims to increase transparency in nursing home ownership by collecting more information on facility owners and their affiliations.
“Our focus is on advancing implementable solutions that promote safe, high-quality care for residents and consider the challenging circumstances some long-term care facilities face,” the statement reads. “We believe the proposed requirements are achievable and necessary.”
CMS is slated to implement the disclosure rules in the fall and release the new data to the public later this year.
“We support transparency and accountability,” the American Health Care Association/National Center for Assisted Living, a trade organization representing the nursing home industry, wrote in response to The Conversation‘s request for comment. “But neither ownership nor line items on a budget sheet prove whether a nursing home is committed to its residents. Over the decades, we’ve found that strong organizations tend to have supportive and trusted leadership as well as a staff culture that empowers frontline caregivers to think critically and solve problems. These characteristics are not unique to a specific type or size of provider.”
It often takes years to improve a poor nursing home − or run one into the ground. The analysis of midsize chains shows that most owners have been associated with their current facilities for less than eight years, making it difficult to separate operators who have taken long-term investments in resident care from those who are looking to quickly extract money and resources before closing them down or moving on. These chains control roughly 41% of nursing home beds in the U.S., according to CMS’s provider data, making the lack of transparency especially ripe for abuse.
A churn of nursing home purchases even during the COVID-19 pandemic shows that investors view the sector as highly profitable, especially when staffing costs are kept low and fines for poor care can easily be covered by the money extracted from residents, their families and taxpayers.
“This is the model of their care: They come in, they understaff and they make their money,” said Sam Brooks, director of public policy at the Consumer Voice, a national resident advocacy organization. “Then they multiply it over a series of different facilities.”

Investor race
The explosion of a billion-dollar private marketplace found its beginnings in government spending.
The adoption of Medicare and Medicaid in 1965 set loose a race among investors to load up on nursing homes, with a surge in for-profit homes gaining momentum because of a reliable stream of government payouts. By 1972, a mere seven years after the inception of the programs, a whopping 106 companies had rushed to Wall Street to sell shares in nursing home companies. And little wonder: They pulled in profits through their ownership of 18% of the industry’s beds, securing about a third of the hefty $3.2 billion of government cash.
The 1990s saw substantial expansion in for-profit nursing home chains, marked by a wave of acquisitions and mergers. At the same time, increasing difficulties emerged in the model for publicly traded chains. Shareholders increasingly demanded rapid growth, and researchers have found that the publicly traded chains tried to appease that hunger by reducing nursing staff and cutting corners on other measures meant to improve quality and safety.
“I began to suspect a possibly inherent contradiction between publicly traded and other large investor-operated nursing home companies and the prerequisites for quality care,” Paul R. Willging, former chief lobbyist for the industry, wrote in a 2007 letter to the editor of The New York Times. “For many investors … earnings growth, quarter after quarter, is often paramount. Long-term investments in quality can work at cross purposes with a mandate for an unending progression of favorable earnings reports.”
One example of that clash can be found at the Ensign Group, founded in 1999 as a private chain of five facilities. Using a strategy of acquiring struggling nursing homes, the company went public in 2007 with more than 60 facilities. What followed was a year-after-year acquisition binge and a track record of growing profits almost every year. Yet the company kept staffing levels below the national average and levels recommended by experts. Its facilities had higher than average inspection deficiencies and higher COVID infection rates. Since 2021, it has racked up more than $6.5 million in penalties.
Ensign did not respond to requests for comment.
Even with that kind of expense cutting, not all publicly traded nursing homes survived as the costs of providing poor care added up. Residents sued over mistreatment. Legal fees and settlements ate into profits, shareholders grumbled, and executives searched for a way out of this Catch-22.
Recognizing the long-term potential for profit growth, private investors snapped up publicly traded for-profit chains, reducing the previous levels of public transparency and oversight. Between 2000 and 2017, 1,674 nursing homes were acquired by private-equity firms in 128 unique deals out of 18,485 facilities. But the same poor-quality problems persisted. Research shows that after snagging a big chain, private investors tended to follow the same playbook: They rebrand the company, increase corporate control and dump unprofitable homes to other investment groups willing to take shortcuts for profit.
Multiple academic studies show the results, highlighting the lower staffing and quality in for-profit homes compared with nonprofits and government-run facilities. Elderly residents staying long term in nursing homes owned by private investment groups experienced a significant uptick in trips to the emergency department and hospitalizations between 2013 and 2017, translating into higher costs for Medicare.
Overall, private-equity investors wreak havoc on nursing homes, slashing registered nurse hours per resident day by 12%, outpacing other for-profit facilities. The aftermath is grim, with a daunting 14% surge in the deficiency score index, a standardized metric for determining issues with facilities, according to a U.S. Department of Health and Human Services report.
The human toll comes in death and suffering. A study updated in 2023 by the National Bureau of Economic Research calculated that 22,500 additional deaths over a 12-year span were attributable to private-equity ownership, equating to about 172,400 lost life years. The calculations also showed that private-equity ownership was responsible for a 6.2% reduction in mobility, an 8.5% increase in ulcer development and a 10.5% uptick in pain intensity.
Hiding in complexity
Exposing the identities of who should be held responsible for such anguish poses a formidable task. Private investors in nursing home chains often employ a convoluted system of limited liability corporations, related companies and family relationships to obscure who controls the nursing homes.
These adjustments are crafted to minimize liability, capitalize on favorable tax policies, diminish regulatory scrutiny and disguise nursing home profitability. In this investigation, entities at every level of involvement with a nursing home denied ownership, even though the same people controlled each organization.
A rule put in place in 2023 by the Centers for Medicare & Medicaid Services requires the identification of all private-equity and real estate investment trust investors in a facility and the release of all related party names. But this hasn’t been enough to surface the players and relationships. More than half of ownership data provided to CMS is incomplete across all facilities, according to a March 2024 analysis of the newly released data.

Even the land under the nursing home is often owned by someone else. In 2021, publicly traded or private real estate investment trusts held a sizable chunk of the approximately $120 billion of nursing home real estate. As with homes owned by private-equity investors, quality measures collapse after REITs get involved, with facilities witnessing a 7% decline in registered nurses’ hours per resident day and an alarming 14% ascent in the deficiency score index. It’s a blatant pattern of disruption, leaving facilities and care standards in a dire state.
Part of that quality collapse comes from the way these investment entities make their money. REITs and their owners can drain cash out of the nursing homes in a number of different ways. The standard tactic for grabbing the money is known as a triple-net lease, where the REIT buys the property then leases it back to the nursing home, often at exorbitant rates. Although the nursing home then lacks possession of the property, it still gets slammed with costs typically shouldered by an owner − real estate taxes, insurance, maintenance and more. Topping it off, the facilities then must typically pay annual rent hikes.
A second tactic that REITs use involves a contracting façade that serves no purpose other than enriching the owners of the trusts. Since triple-net lease agreements prohibit REITs from taking profits from operating the facilities, the investors create a subsidiary to get past that hurdle. The subsidiary then contracts with a nursing home operator − often owned or controlled by another related party − and then demands a fee for providing operational guidance. The use of REITs for near-risk-free profits from nursing homes has proven to be an ever-growing technique, and the midsize chains, which our investigation found generally provided the worst care, grew in their reliance on REITs during the pandemic.
“When these REITs start coming in … nursing homes are saddled with these enormous rents, and then they wind up going out of business,” said Richard Mollot, executive director of the Long-Term Care Community Coalition, a nonprofit organization that advocates for better care at nursing homes. “It’s no longer a viable facility.”
The churn of nursing home purchases by midsize chains underscores investors’ perception of the sector’s profitability, particularly when staffing expenses are minimized and penalties for subpar care can be offset by money extracted through related transactions and payments from residents, their families and taxpayers. Lawsuits can drag out over years, and in the worst case, if a facility is forced to close, its land and other assets can be sold to minimize the financial loss.
Take Brius Healthcare, a name that resonates with a disturbing cadence in the world of nursing home ownership. A search of the federal database for nursing home ownership and penalties shows that Brius was responsible for 32 facilities as of the start of 2024, but the true number is closer to 80, according to BriusWatch.org, which tracks violations. At the helm of this still midsize network stands Shlomo Rechnitz, who became a billionaire in part by siphoning from government payments to his facilities scattered across California, according to a federal and state lawsuit.
In lawsuits and regulators’ criticisms, Rechnitz’s homes have been associated with tales of abuse, as well as several lawsuits alleging terrible care. The track record was so bad that, in the summer of 2014, then-California Attorney General Kamala Harris filed an emergency motion to block Rechnitz from acquiring 19 facilities, writing that he was “a serial violator of rules within the skilled nursing industry” and was “not qualified to assume such an important role.”
Yet, Rechnitz’s empire in California surged forward, scooping up more facilities that drained hundreds of millions of federal and state funds as they racked up pain and profit. The narrative played out at Windsor Redding Care Center in Redding, California. Rechnitz bought it from a competing nursing home chain and attempted to obtain a license to operate the facility. But in 2016, the California Department of Public Health refused the application, citing a staggering 265 federal regulatory violations across his other nursing homes over just three years.
According to court filings, Rechnitz formed a joint venture with other investors who in turn held the license. Rechnitz, through the Brius joint venture, became the unlicensed owner and operator of Windsor Redding.
Brius carved away at expenses, slashing staff and other care necessities, according to a 2022 California lawsuit. One resident was left to sit in her urine and feces for hours at a time. Overwhelmed staff often did not respond to her call light, so once she instead climbed out of bed unassisted, fell and fractured her hip. Other negligence led to pressure ulcers, and when she was finally transferred to a hospital, she was suffering from sepsis. She was not alone in her suffering. Numerous other residents experienced an unrelenting litany of injuries and illnesses, including pressure ulcers, urinary tract infections from poor hygiene, falls, and skin damage from excess moisture, according to the lawsuit.
In 2023, California moved forward with licensing two dozen of Rechnitz’s facilities with an agreement that included a two-year monitoring period, right before statewide reforms were set to take effect. The reforms don’t prevent existing owners like Rechnitz from continuing to run a nursing home without a license, but they do prevent new operators from doing so.
“We’re seeing more of that, I think, where you have a proliferation of really bad operators that keep being provided homes,” said Brooks, the director of public policy at the Consumer Voice. “There’s just so much money to be made here for unscrupulous people, and it just happens all the time.”
Rechnitz did not respond to multiple requests for comment. Bruis also did not respond.
Perhaps no other chain showcases the havoc that can be caused by one individual’s acquisition of multiple nursing homes than Skyline Health Care. The company’s owner, Joseph Schwartz, parlayed the sale of his insurance business into ownership of 90 facilities between mid-2016 and December 2017, according to a federal indictment. He ran the company out of an office above a New Jersey pizzeria and at its peak managed facilities in 11 states.
Schwartz went all-in on cost cutting, and by early 2018, residents were suffering from the shortage of staff. The company wasn’t paying its bills or its workers. More than a dozen lawsuits piled up. Last year, Schwartz was arrested and faced charges in federal district court in New Jersey for his role in a $38 million payroll tax scheme. In 2024, Schwartz pleaded guilty to his role in the fraud scheme. He is awaiting sentencing, where he faces a year in prison along with paying at least $5 million in restitution.
Skyline collapsed and disrupted thousands of lives. Some states took over facilities; others closed, forcing residents to relocate and throwing families into chaos. The case also highlights the ease with which some bad operators can snap up nursing homes with little difficulty, with federal and state governments allowing ownership changes with little or no review.
Schwartz’s lawyer did not respond to requests for comment.
Not that nursing homes have much to fear in the public perception of their reputation for quality. CMS uses what is known as the Five-Star Quality Rating System, designed to help consumers compare nursing homes to find one that provides good care. Theoretically, nursing homes with five-star ratings are supposed to be exceptional, while those with one-star ratings are deemed the worst. But research shows that nursing homes can game the system, with the result that a top star rating might reflect little more than a facility’s willingness to cheat.
A star rating is composed of three parts: The score from a government inspection and the facility’s self-reports of staffing and quality. This means that what the nursing homes say about themselves can boost the star rating of facilities even if they have poor inspection results.
Multiple studies have highlighted a concerning trend: Some nursing homes, especially for-profit ones, inflate their self-reported measures, resulting in a disconnect from actual inspection findings. Notably, research suggests that for-profit nursing homes, driven by significant financial motives, are more likely to engage in this practice of inflating their self-reported assessments.
At bottom, the elderly and their families seeking quality care unknowingly find themselves in an impossible situation with for-profit nursing homes: Those facilities tend to provide the worst quality, and the only measure available for consumers to determine where they will be treated well can be rigged. The result is the transformation of an industry meant to care for the most vulnerable into a profit-driven circus.

The pandemic
Nothing more clearly exposed the problems rampant in nursing homes than the pandemic. Throughout that time, nursing homes reported that almost 2 million residents had infections and 170,000 died.
No one should have been surprised by the mass death in nursing homes − the warning signs of what was to come had been visible for years. Between 2013 and 2017, infection control was the most frequently cited deficiency in nursing homes, with 40% of facilities cited each year and 82% cited at least once in the five-year period. Almost half were cited over multiple consecutive years for these deficiencies − if fixed, one of the big causes of the widespread transmission of COVID in these facilities would have been eliminated.
But shortly after coming into office in 2017, the Trump administration weakened what was already a deteriorating system to regulate nursing homes. The administration directed regulators to issue one-time fines against nursing homes for violations of federal rules rather than for the full time they were out of compliance. This shift meant that even nursing homes with severe infractions lasting weeks were exempted from fines surpassing the maximum per-instance penalty of $20,965.
Even that near-worthless level of regulation was not feeble enough for the industry, so lobbyists pressed for less. In response, just a few months before COVID emerged in China, the Trump administration implemented new regulations that effectively abolished a mandate for each to hire a full-time infection control expert, instead recommending outside consultants for the job.
The perfect storm had been reached, with no experts required to be on site, prepared to combat any infection outbreaks. On Jan. 20, 2020 − just 186 days after the change in rules on infection control − the CDC reported that the first laboratory-confirmed case of COVID had been found at a nursing home in Washington state.
The least prepared in this explosion of disease were the for-profit nursing homes, compared with nonprofit and government facilities. Research from the University of California at San Francisco found those facilities were linked to higher numbers of COVID cases. For-profits not only had fewer nurses on staff but also high numbers of infection-control deficiencies and lower compliance with health regulations.
Even as the United States went through the crisis, some owners of midsize chains continued snapping up nursing homes. For example, two Brooklyn businessmen named Simcha Hyman and Naftali Zanziper were going on a nursing home buying spree through their private-equity company, the Portopiccolo Group. Despite poor ratings in their previously owned facilities, nothing blocked the acquisitions.
One such facility was a struggling nursing home in North Carolina now known as The Citadel Salisbury. Following the traditional pattern forged by private investors in the industry, the new owners set up a convoluted network of business entities and then used them to charge the nursing home for services and property. A 2021 federal lawsuit of many plaintiffs claimed that they deliberately kept the facility understaffed and undersupplied to maximize profit.
Within months of the first case of COVID reported in America, The Citadel Salisbury experienced the largest nursing home outbreak in the state. The situation was so dire that on April 20, 2020, the local medical director of the emergency room took to the local newspaper to express his distress, revealing that he had pressed the facility’s leadership and the local health department to address the known shortcomings.
The situation was “a blueprint for exactly what not to do in a crisis,” medical director John Bream wrote. “Patients died at the Citadel without family members being notified. Families were denied the ability to have one last meaningful interaction with their family. Employees were wrongly denied personal protective equipment. There has been no transparency.”
After a series of scathing inspection reports, the facility finally closed in the spring of 2022. As for the federal lawsuit, court documents show that a tentative agreement was reached in 2023. But the case dragged out for nearly three years, and one of the plaintiffs, Sybil Rummage, died while seeking accountability through the court.
Still, the pandemic had been a time of great success for Hyman and Zanziper. At the end of 2020, they owned more than 70 facilities. By 2021, their portfolio had exploded to more than 120. Now, according to data from the Centers for Medicare & Medicaid Services, Hyman and Zanziper are associated with at least 131 facilities and have the highest amount of total fines recorded by the agency for affiliated entities, totaling nearly $12 million since 2021. And their average fine per facility, as calculated by CMS, is more than twice the national average at almost $90,000.
In a written statement, Portopiccolo Group spokesperson John Collins disputed that the facilities had skimped on care and argued that they were not managed by the firm. “We hire experienced, local health care teams who are in charge of making all on-the-ground decisions and are committed to putting residents first.” He added that the number of facilities given by CMS was inaccurate but declined to say how many are connected to its network of affiliates or owned by Hyman and Zanziper.
With the nearly 170,000 resident deaths from COVID and many related fatalities from isolation and neglect in nursing homes, in February 2022 President Biden announced an initiative aimed at improving the industry. In addition to promising to set a minimum staffing standard, the initiative is focused on improving ownership and financial transparency.
“As Wall Street firms take over more nursing homes, quality in those homes has gone down and costs have gone up. That ends on my watch,” Biden said during his 2022 State of the Union address. “Medicare is going to set higher standards for nursing homes and make sure your loved ones get the care they deserve and expect.”

Still, the current trajectory of actions appears to fall short of what’s needed. While penalties against facilities have sharply increased under Biden, some of the Trump administration’s weak regulations have not been replaced.
A rule proposed by CMS in September 2023 and released for review in March 2024 would require states to report what percentage of Medicaid funding is used to pay direct care workers and support staff and would require an RN on duty 24/7. It would also require a minimum of three hours of skilled staffing care per patient per day. But the three-hour minimum is substantially lower than the 4.1 hours of skilled staffing for nursing home residents suggested by CMS over two decades ago.
The requirements are also lower than the 3.8 average nursing staff hours already employed by U.S. facilities.
The current administration has also let stand the Trump administration reversal of an Obama rule that banned binding arbitration agreements in nursing homes.
It breaks a village
The Villages of Orleans Health and Rehabilitation Center in Albion, New York, was, by any reasonable measure, broken. Court records show that on some days there was no nurse and no medication for the more than 100 elderly residents. Underpaid staff spent their own cash for soap to keep residents clean. At times, the home didn’t feed its frail occupants.
Meanwhile, according to a 2022 lawsuit filed by the New York attorney general, riches were siphoned out of the nursing home and into the pockets of the official owner, Bernard Fuchs, as well as assorted friends, business associates and family. The lawsuit says $18.7 million flowed from the facility to entities owned by a group of men who controlled the Village’s operations.
Although these men own various nursing homes, Medicare records show few connections between them, despite them all being investors in Comprehensive Healthcare Management, which provided administrative services to the Villages. Either they or their families were also owners of Telegraph Realty, which leased what was once the Villages’ own property back to the facility at rates the New York attorney general deemed exorbitant, predatory and a sham.
So it goes in the world of nursing home ownership, where overlapping entities and investors obscure the interrelationships between them to such a degree that Medicare itself is never quite sure who owns what.
Glenn Jones, a lawyer representing Comprehensive Healthcare Management, declined to comment on the pending litigation, but he forwarded a court document his law firm filed that labels the allegations brought by the New York attorney general “unfounded” and reliant on “a mere fraction” of its residents.

The shadowy structure of ownership and related party transactions plays an enormous role in how investors enrich themselves, even as the nursing homes they control struggle financially. Compounding the issue, the figures reported by nursing homes regarding payments to related parties frequently diverge from the disclosures made by the related parties themselves.
As an illustration of the problems, consider Pruitt Health, a midsize chain with 87 nursing homes spread across Georgia, South Carolina, North Carolina and Florida that had low overall federal quality ratings and about $2 million in penalties. A report by The National Consumer Voice For Quality Long-Term Care, a consumer advocacy group, shows that Pruitt disclosed general related party costs nearing $482 million from 2018 to 2020. Yet in that same time frame, Pruitt reported payments to specific related parties amounting to about $570 million, indicating a $90 million excess. Its federal disclosures offer no explanation for the discrepancy. Meanwhile, the company reported $77 million in overall losses on its homes.
The same pattern holds in the major chains such as the Cleveland, Tennessee-based Life Care Centers of America, which operates roughly 200 nursing homes across 27 states, according to the report. Life Care’s financial disbursements are fed into a diverse spectrum of related entities, including management, staffing, insurance and therapy companies, all firmly under the umbrella of the organization’s ownership. In fiscal year 2018, the financial commitment to these affiliated entities reached $386,449,502; over the three-year period from 2018 to 2020, Life Care’s documented payments to such parties hit an eye-popping $1.25 billion.
Pruitt Health and Life Care Centers did not respond to requests for comment.
Overall, 77% of US nursing homes reported $11 billion in related-party transactions in 2019 − nearly 10% of total net revenues − but the data is unaudited and unverified. The facilities are not required to provide any details of what specific services were provided by the related parties, or what were the specific profits and administrative costs, creating a lack of transparency regarding expenses that are ambiguously categorized under generic labels such as “maintenance.” Significantly, there is no mandate to disclose whether any of these costs exceed fair market value.
What that means is that nursing home owners can profit handsomely through related parties even if their facilities are being hit with repeated fines for providing substandard care.
“What we would consider to be a big penalty really doesn’t matter because there’s so much money coming in,” said Mollot of the Long-Term Care Community Coalition. “If the facility fails, so what? It doesn’t matter. They pulled out the resources.’’
Hiding profit
Ultimately, experts say, this ability to drain cash out of nursing homes makes it almost impossible for anyone to assess the profitability of these facilities based on their public financial filings, known as cost reports.
"The profit margins (for nursing homes) also should be taken with a grain of salt in the cost reports,” said Dr. R. Tamara Konetzka, a University of Chicago professor of public health sciences, at a recent meeting of the Medicare Payment Advisory Commission. “If you sell the real estate to a REIT or to some other entity, and you pay sort of inflated rent back to make your profit margins look lower, and then you recoup that profit because it’s a related party, we’re not going to find that in the cost reports.”
That ability to hide profits is key to nursing homes’ ability to block regulations to improve quality of care and to demand greater government payments. For decades, the industry’s refrain has been that cuts in reimbursements or requirements to increase staffing will drive facilities into bankruptcy; already, they claim, half of all nursing homes are teetering on the edge of collapse, the result, they say, of inadequate Medicaid rates. All in all, the industry reports that less than 3% of their revenue goes to earnings.
But that does not include any of the revenue pulled out of the homes to boost profits of related parties controlled by the same owners pleading poverty. And this tactic is only one of several ways that the nursing home industry disguises its true profits, giving it the power to plead poverty to an unknowing government.
Under the regulations, only certain nursing home expenses are reimbursable, such as money spent for care. Many others − unreasonable payments to the headquarters of chains, luxury items, and fees for lobbyists and lawyers − are disallowed after Medicare reviews the cost reports. But by that time, the government has already reimbursed the nursing homes for those expenses − and none of those revenues have to be returned.
Data indicates that owners also profit by overcharging nursing homes for services and leases provided by related entities. A March 2024 study from Lehigh University and the University of California, Los Angeles shows that costs were inflated when nursing home owners changed from independent contractors to businesses owned or controlled directly or indirectly by the same people. Overall, spending on real estate increased 20.4%, and spending on management increased 24.6% when the businesses were affiliated, the research showed.
Nursing homes also claim that noncash depreciation cuts into their profits. Those expenses, which show up only in accounting ledgers, assume that assets such as equipment and facilities are gradually decreasing in value and ultimately will need to be replaced.
That might be reasonable if the chains purchased new items once their value depreciated to zero, but that is not always true. A 2004 report by the Medicare Payment Advisory Commission found that the depreciation claimed by health care companies, including nursing homes, may not reflect actual capital expenditures or the actual market value.
If disallowed expenses and noncash depreciation were not included, profit margins for the nursing home industry would jump to 8.8%, far more than the 3% it claims. And given that these numbers all come from nursing home cost reports submitted to the government, they may underestimate the profits even more. Audited cost reports are not required, and the Government Accountability Office has found that CMS does little to ensure the numbers are correct and complete.
This lack of basic oversight essentially gives dishonest nursing home owners the power to grab more money from Medicare and Medicaid while being empowered to claim that their financials prove they need more.
“They face no repercussions,” Brooks of Consumer Voice said, commenting on the current state of nursing home operations and their unscrupulous owners. “That’s why these people are here. It’s a bonanza to them.”
Ultimately, experts say, finding ways to force nursing homes to provide quality care has remained elusive. Michael Gelder, former senior health policy adviser to then-Gov. Pat Quinn of Illinois, learned that brutal lesson in 2010 as head of a task force formed by Quinn to investigate nursing home quality. That group successfully pushed a new law, but Gelder now says his success failed to protect this country’s most vulnerable citizens.
“I was perhaps naively convinced that someone like myself being in the right place at the right time with enough resources could really fix this problem,” he said. “I think we did the absolute best we could, and the best that had ever been done in modern history up to that point. But it wasn’t enough. It’s a battle every generation has to fight.”
Click here to learn more about how some existing tools can address problems with for-profit nursing homes.
Sean Campbell is an adjunct assistant professor at Columbia University and a contributing writer at the Garrison Project, an independent news organization that focuses on mass incarceration and criminal justice.
Harrington is an advisory board member of the nonprofit Veteran's Health Policy Institute and a board member of the nonprofit Center for Health Information and Policy. Harrington served as an expert witness on nursing home litigation cases by residents against facilities owned or operated by Brius and Shlomo Rechnitz in the past and in 2022. She also served as an expert witness in a case against The Citadel Salisbury in North Carolina in 2021.
bankruptcy pandemic covid-19 fed reit real estate cdc secretary of health trump medication therapy spread deaths transmission china

Net Zero, The Digital Panopticon, & The Future Of Food


Riley Gaines Explains How Women’s Sports Are Rigged To Promote The Trans Agenda


Health Officials: Man Dies From Bubonic Plague In New Mexico


MIPIM 2024 Reflects Mixed Feelings on CRE Recovery


Five Aerospace Investments to Buy as Wars Worsen Copy


Trump nearly derailed democracy once − here’s what to watch out for in reelection campaign


For-profit nursing homes are cutting corners on safety and draining resources with financial shenanigans − especially at midsize chains that dodge public scrutiny


Analyst reviews Apple stock price target amid challenges


Association of prenatal vitamins and metals with epigenetic aging at birth and in childhood


Stock Market Today: Stocks turn lower as factory inflation spikes, retail sales miss target
-

 Uncategorized3 weeks ago
Uncategorized3 weeks agoAll Of The Elements Are In Place For An Economic Crisis Of Staggering Proportions
-

 International6 days ago
International6 days agoEyePoint poaches medical chief from Apellis; Sandoz CFO, longtime BioNTech exec to retire
-

 Uncategorized4 weeks ago
Uncategorized4 weeks agoCalifornia Counties Could Be Forced To Pay $300 Million To Cover COVID-Era Program
-

 Uncategorized3 weeks ago
Uncategorized3 weeks agoApparel Retailer Express Moving Toward Bankruptcy
-

 Uncategorized4 weeks ago
Uncategorized4 weeks agoIndustrial Production Decreased 0.1% in January
-

 International6 days ago
International6 days agoWalmart launches clever answer to Target’s new membership program
-

 Uncategorized4 weeks ago
Uncategorized4 weeks agoRFK Jr: The Wuhan Cover-Up & The Rise Of The Biowarfare-Industrial Complex
-

 Uncategorized3 weeks ago
Uncategorized3 weeks agoGOP Efforts To Shore Up Election Security In Swing States Face Challenges