Government
Gilead 2020: HIV … and COVID?
Gilead 2020: HIV … and COVID?
Share this:


HIV products account for a considerable majority of Gilead’s revenue, though one oncologic is beginning to creep up the sales ladder and an experimental drug is showing signs of effectiveness against COVID-19.
By Joshua Slatko • josh.slatko@medadnews.com
FINANCIAL PERFORMANCE
(All figures are in millions of dollars, except EPS)
2019
Revenue $22,449
Net income $5,364
Diluted EPS $4.22
R&D expense $9,106
1H 2020
Revenue $10,691
Net income $(1,808)
Diluted EPS $(1.42)
R&D expense $2,303
BEST-SELLING Rx PRODUCTS
(All sales are in millions of dollars)
2019
Biktarvy $4,738
Genvoya $3,931
Truvada $2,813
Sofosbuvir/Velpatasvir $1,965
Odefsey $1,655
Descovy $1,500
Ledipasvir/Sofosbuvir $643 $
Letairis $618
Atripla $600
1H 2020
Biktarvy $3,297
Genvoya $1,640
Sofosbuvir/Velpatasvir $899
Descovy $875
Truvada $793
Odefsey $791
Yescarta $296
Vemlidy $287
Outcomes Creativity Index Score: 9
Manny Awards – N/A
Cannes Lions – N/A
LIA: Health & Wellness – N/A
Clio Health – N/A
One Show: HW&P – N/A
MM&M Awards – 3
Global Awards – 2
Creative Floor Awards – 4
HIV has always been a particular specialty at Gilead, from the company’s in-licensing of tenofovir (later Viread) in 1991 all the way through to the launch of Biktarvy in 2018 and beyond. But with the decline of the company’s hepatitis C blockbusters Sovaldi, Harvoni and Epclusa, Gilead is depending on its HIV portfolio more than ever. In 2019 the company’s HIV products accounted for just short of 75 percent of its total revenue; in the first half of 2020 the ratio was even higher, just over 77 percent. Of course, being the go-to company for HIV is not so bad; Gilead’s HIV sales have grown at a 12 percent per year clip over the past three years, while the company’s sales of other products have bounced up and down.

“As we accumulate more data from global clinical trials and initiate many additional studies, we will understand more about the full value of remdesivir over time,” says Gilead CEO Daniel O’Day.
Of course HIV is not all there is to Gilead. The company’s lymphoma drug Yescarta has generated impressive sales growth in 2020 and recently earned a Breakthrough Therapy designation from FDA for additional indications. And remdesivir – in its previous life a failed experimental drug for hepatitis C, Ebola, and Marburg virus – is showing signs of effectiveness against severe cases of COVID-19, enough so that FDA granted the drug an Emergency Use Authorization under the trade name Veklury.
“Our work on remdesivir is far from done,” says Gilead CEO Daniel O’Day. “We continue to explore its potential to help in this pandemic in various ways, such as evaluating treatment earlier in the course of the disease, in outpatient settings, with an inhaled formulation, in additional patient groups and in combination with other therapies. As we accumulate more data from global clinical trials and initiate many additional studies, we will understand more about the full value of remdesivir over time. Our teams also remain focused on increasing supplies to meet the high global demand. By the end of this year, we expect our investment on the development and manufacture of remdesivir to exceed $1 billion and our commitment will continue through 2021 and beyond.”
Gilead’s top-line revenue was $22.45 billion in 2019, edging up 1.5 percent from the previous year. Net income declined by 1.8 percent to $5.36 billion, while diluted earnings per share were up five cents to $4.22. In the first half of 2020, top-line revenue declined 2.5 percent to $10.69 billion while net income fell into the red at negative $1.81 billion and a loss of $1.42 per share. However, this was impacted by a $4.5 billion charge for in-process R&D related to the acquisition of Forty Seven in April. Without this charge and other one-time items, company executives estimate that EPS for the first half of 2020 would have been $2.80. The company is projecting full-year 2020 EPS to fall between $0.83 and $2.23.
Partnerships & acquisitions
Gilead in January announced the licensing of The Rockefeller University’s portfolio of broadly neutralizing antibodies (bNAbs) against HIV, including the two clinical-stage agents 3BNC117 and 10-1074. These investigational agents, company executives say, have potential for use in HIV long-acting therapies for treatment and prevention, as well as cure strategies.
HIV bNAbs are a class of immunotherapy agents that were originally derived from HIV-infected individuals with a strong anti-HIV immunologic response and were designed to target HIV, particularly originating from the latent viral reservoir. Initial pre-clinical and clinical research has shown that HIV bNAbs can produce an enhanced, prolonged immune response to HIV, representing a promising new approach for HIV treatment or prevention in combination with other long-acting agents, or prolonged virologic remission in the absence of antiretroviral use. This licensing agreement provides an additional pathway for innovations in Gilead’s HIV pipeline in areas of evolving and unmet medical need.
Under the agreement, Gilead acquired exclusive global licenses to develop and commercialize Rockefeller’s full portfolio of HIV bNAbs. Rockefeller received an upfront payment and is eligible to receive cumulative milestone payments, as well as royalties on net sales. Additionally, Rockefeller will retain rights to perform non-clinical and early-stage clinical research on the portfolio of HIV antibodies.
In March, Gilead and Forty Seven Inc. entered into a definitive agreement pursuant to which Gilead would acquire Forty Seven for $95.50 per share in cash. The transaction, which valued Forty Seven at about $4.9 billion, closed the following month.
Through the addition of Forty Seven’s investigational lead product candidate, magrolimab, company leaders expect the acquisition to strengthen Gilead’s immuno-oncology research and development portfolio. Magrolimab is a monoclonal antibody in clinical development for the treatment of several cancers for which new, transformative medicines are urgently needed, including myelodysplastic syndrome, acute myeloid leukemia, and diffuse large B-cell lymphoma. The investigational therapy targets CD47, a “do not eat me” signal that allows cancer cells to avoid destruction thereby permitting the patient’s own innate immune system to engulf and eradicate those cancer cells. Forty Seven presented promising results of a Phase Ib study of magrolimab in patients with MDS and AML at the American Society of Hematology meeting in December 2019. Magrolimab has the potential to be a first-in-class therapy.
In April, Gilead subsidiary Kite and oNKo-innate announced the companies had entered into a three-year cancer immunotherapy research collaboration to support discovery and development of next-generation drug and engineered cell therapies focused on natural killer cells.
Current cancer immunotherapy approaches primarily focus on T cell mediated anti-tumor immunity, including checkpoint inhibition, and chimeric antigen receptor (CAR) T cell therapy. Like T cells, NK cells are a class of lymphocytes (white blood cells) that play a critical surveillance and effector role in the immune system. NK cells and T cells each have the potential to attack cancer cells, but have different mechanisms for tumor cell killing. Thus, appropriately activated and targeted NK cells may represent a differentiated approach that would be potentially complementary and synergistic with T cell mediated anti-tumor strategies.
Through this research collaboration, oNKo-innate will use genome-wide screening techniques and its proprietary technology platform to discover novel immune cell targets that enhance NK cell anti-tumor immunity and to create NK cell therapies. For Gilead, oNKo-innate will execute screens to identify and validate targets to seed internal Gilead immuno-oncology discovery programs. For Kite, oNKo-innate will create and evaluate NK constructs for Kite’s development of next-generation cell therapies.
Also in April, Gilead and Second Genome, a microbiome science company, announced a four-year strategic collaboration to identify biomarkers associated with clinical response in up to five of Gilead’s pipeline compounds in inflammation, fibrosis and other diseases, and to identify potential new targets and drug candidates for the treatment of inflammatory bowel disease.
Under the terms of the agreement, Second Genome will utilize its proprietary Microbiome Analytics Platform to identify novel biomarkers associated with clinical response to Gilead’s investigational medicines. This work will harness the latest insights in microbiome science to help inform patient stratification and optimize potential treatments for patients in the future. The platform, in combination with additional discovery and development tools, will also seek to identify new targets and drug candidates relevant to IBD. This will include the identification of up to five novel IBD targets or drug candidates over the next four years, with an option to extend the collaboration for an additional two years.
Also in April, Kite and Teneobio Inc. entered into a license and collaboration agreement through which Kite will receive exclusive rights to certain antibodies directed to B-cell maturation antigen. The fully human variable heavy chain of one such antibody is undergoing clinical evaluation in a chimeric antigen receptor format for the treatment of patients with multiple myeloma in a Phase 1 clinical trial at the National Cancer Institute. Kite and Teneobio will also collaborate on the discovery of antibodies directed to four additional targets, using Teneobio’s proprietary Human Heavy-Chain Antibodies (UniAb) platform, to be used in CAR T cell therapies for multiple myeloma and other cancers.
CARs traditionally utilize an antigen-recognition domain made up of a single chain variable fragment (scFv). The binding domain derived from Teneobio’s BCMA antibody has key potential benefits, including minimal potential for immunogenicity due to its lack of murine elements, and its small size, which may allow for the creation of dual-targeting CAR T therapies.
Under the terms of the agreement, Teneobio received an upfront payment and will be eligible to receive additional payments based on achievement of certain clinical and regulatory milestones, as well as royalties on future potential sales.
In June, Gilead announced that for $275 million the company would acquire a 49.9 percent equity interest in Pionyr Immunotherapeutics Inc., a privately held company developing first-in-class cancer immunotherapies, and an exclusive option to purchase the remainder of Pionyr. Under the agreement, Pionyr’s shareholders may receive up to an additional $1.47 billion in option exercise fees and future milestone payments.
Pionyr’s Myeloid Tuning therapies have the potential to treat patients who currently do not benefit from checkpoint inhibitor therapies. PY314 and PY159 have demonstrated preclinical efficacy, suggesting potential in solid tumors in combination with established anti-PD(L)-1 agents. Pionyr plans to file investigational new drug applications with FDA for both PY314 and PY159 in the third quarter of this year. Pending Phase Ib results from either candidate – or sooner if Gilead chooses – Gilead can exercise its exclusive option to acquire the remainder of Pionyr.
Under the terms of the agreement, Pionyr’s shareholders will receive $275 million upon closing. Gilead will receive 49.9 percent of the common stock of Pionyr and an exclusive option to purchase the remaining equity. Gilead may exercise the company’s exclusive option upon completion of Phase 1b studies for PY314 and PY159, or at an earlier time if Gilead chooses to do so, for a $315 million option exercise fee and up to $1.15 billion in potential future milestone payments. In addition, Gilead will provide Pionyr with additional funding for the PY314 and PY159 clinical programs, as well as ongoing research and development programs.
In July, Gilead announced that it would invest $300 million to acquire a 49.9 percent equity interest in Tizona Therapeutics Inc., a privately held company developing first-in-class cancer immunotherapies. Gilead also received an exclusive option to acquire the remainder of Tizona for up to an additional $1.25 billion, including an option exercise fee and potential future milestone payments.
Gilead can exercise its option to acquire the remainder of Tizona following the readout of a Phase Ib study of Tizona’s investigational antibody, TTX-080, or earlier if Gilead decides to do so. TTX-080, discovered by Tizona, is a potential first-in-class medicine that targets HLA-G, a novel and emerging immune checkpoint expressed across multiple tumor types. The expression pattern of HLA-G often appears distinct from that of PD-(L)1, suggesting potential utility to address tumors that do not respond to current anti-PD-(L)1 treatments and to deepen responses in tumors that are sensitive to anti-PD-(L)1 therapies. FDA has cleared Tizona’s investigational new drug application for TTX-080, and in the third quarter of 2020, Tizona planned to initiate a Phase I clinical trial evaluating TTX-080 both as a monotherapy and in combination with other agents in patients with advanced cancers.
Under the terms of the agreement, Tizona equity holders received $300 million upon closing. Gilead obtained a 49.9 percent equity stake and an exclusive option to purchase the remaining equity exercisable following the completion of Phase Ib studies for TTX-080, or earlier if Gilead chooses. Tizona equity holders are eligible to receive up to $1.25 billion in an option exercise fee and potential future milestone payments. Gilead will also provide funding to support Tizona’s ongoing research and development to advance its novel pipeline. Tizona will spin off TTX-030, the company’s investigational, first-in-class anti-CD39 antibody partnered with AbbVie, into a separate entity prior to closing of this transaction. TTX-030 is not subject to this agreement.
Also in July, Gilead and Arcus Biosciences Inc., an oncology-focused biopharmaceutical company working to create best-in-class cancer therapeutics, announced the closing of their option and co-development and co-commercialization partnership agreement signed in May. Under the terms of the agreements, the closing of this transaction triggered a payment of $175 million by Gilead to Arcus. In addition, Gilead made an equity investment in Arcus of about $200 million by purchasing shares at a price of $33.54 per share. As a result of this investment and Gilead’s participation in Arcus’s follow-on offering in May, Gilead now owns nearly 8.2 million shares of common stock of Arcus, representing about 13 percent of Arcus’s outstanding shares.
Arcus Biosciences is an oncology-focused biopharmaceutical company taking advantage of its cross-disciplinary expertise to discover highly differentiated therapies and to develop a broad portfolio of novel combinations addressing significant unmet needs. Arcus has four molecules in clinical development. AB928, the first dual A2a/A2b adenosine receptor antagonist in the clinic, is being evaluated in multiple Phase Ib/II studies across different indications, including prostate, colorectal, non-small cell lung, pancreatic, triple-negative breast, and renal cell cancers. AB680, the first small-molecule CD73 inhibitor in the clinic, is in Phase 1 development for first-line treatment of metastatic pancreatic cancer in combination with zimberelimab and gemcitabine/nab-paclitaxel. AB154, an anti-TIGIT monoclonal antibody and new potential immuno-oncology backbone therapy, is in a three-arm randomized Phase II study for first-line treatment of PD-L1-high metastatic non-small cell lung cancer evaluating zimberelimab monotherapy, AB154 with zimberelimab and AB154 plus AB928 with zimberelimab. Zimberelimab (AB122), Arcus’s anti-PD-1 monoclonal antibody, is also being evaluated in a Phase Ib study as monotherapy for cancers with no approved anti-PD-1 treatment options, and in various combinations across the portfolio.
In August, Gilead and Tango Therapeutics announced an expanded strategic collaboration focused on the discovery, development and commercialization of innovative targeted immune evasion therapies for patients with cancer. Under the expanded multi-year collaboration, which builds on an agreement signed in 2018, Tango will continue to take advantage of its proprietary, CRISPR-enabled functional genomics target discovery platform to identify novel immune evasion targets. The number of targets covered will expand from five to 15. Gilead has options to worldwide rights for programs directed at these targets over the next seven years. Gilead also has the right to pay option extension fees for Tango to lead activities through early clinical development, to which Gilead will retain its option rights. Tango has the option to co-develop and co-promote the lead products for up to five programs in the United States.
The collaboration excludes Tango’s lead programs, including one program that is expected to be in investigational new drug application-enabling studies in 2021. Tango also retains the rights to identify targets outside the immune evasion space as the company continues to build its wholly owned pipeline.
Under the terms of the collaboration, Gilead is responsible for a $125 million upfront payment to Tango and a $20 million equity investment in the company. In addition, Gilead has the right to option up to 15 programs over the seven-year collaboration for up to $410 million per program in opt-in, extension and milestone payments. Tango is also eligible to receive up to low double-digit tiered royalties on net sales. For those products that Tango opts to co-develop and co-promote, the parties will equally split profits and losses, as well as development costs, in the United States, and Tango is eligible to receive milestone payments and royalties on ex-U.S. sales.
In September, Gilead announced an agreement with Jounce Therapeutics Inc., a clinical-stage company focused on the discovery and development of novel cancer immunotherapies and predictive biomarkers, to exclusively license its JTX-1811 program.
JTX-1811 is a monoclonal antibody designed to selectively deplete immunosuppressive tumor-infiltrating T regulatory cells. The target of JTX-1811 is CCR8, a chemokine receptor enriched on TITR cells. When JTX-1811 binds to CCR8, it targets TITR cells for depletion by enhanced antibody-dependent cellular cytotoxicity mechanism. The antibody remains on track for filing an investigational new drug application in the first half of 2021.
Under the terms of the agreement, Gilead will make a $85 million upfront payment to, and a $35 million equity investment at a premium in, Jounce upon closing. In addition, Jounce may receive up to an additional $685 million in future clinical, regulatory, and commercial milestone payments. Jounce is additionally eligible to receive royalties ranging from high single digit to mid-teens based upon worldwide sales, subject to certain adjustments.
Jounce will lead development of JTX-1811 through IND clearance, and thereafter, Gilead will have the sole right to develop JTX-1811. JTX-1811 is not approved anywhere globally. Its efficacy and safety have not been established.
Also in September, Kite and HiFiBiO Therapeutics announced the companies have entered into a two-year research collaboration and license agreement in acute myeloid leukemia. Through this collaboration, HiFiBiO is the company’s proprietary technology platforms to identify novel AML targets and anti-AML specific antibodies for Kite’s use in cell therapies.
Under the terms of the agreement, HiFiBiO received an upfront payment and is eligible for additional payments based on the achievement of certain research milestones. Kite has an exclusive option to opt in on any targets discovered through the collaboration, for which HiFiBiO will receive an additional payment and is eligible for additional development, regulatory and commercial milestone payments, as well as royalty payments.
Product performances
The one-pill once-a-day FTC/TAF HIV product Biktarvy leaped to the top of Gilead’s portfolio in 2019, with sales more than quadrupling to $4.74 billion, moving the medicine past stable-mate Genvoya to become the world’s leading HIV brand by sales. Biktarvy’s growth has continued impressively in 2020, with sales up another 72.7 percent to $3.3 billion in the year’s first half.
In March, Gilead announced data from the BRAAVE 2020 study, a Phase III clinical trial evaluating the safety and efficacy of switching to Biktarvy in virologically suppressed adults living with HIV who self-identified as Black or African American. The data showed that, at 24 weeks, switching to Biktarvy from a standard regimen of two nucleoside reverse transcriptase inhibitors plus a third agent may potentially be an effective and well-tolerated treatment regimen in patients with a history of treatment failure or pre-existing resistance, and did not result in treatment emergent resistance to study drugs with Biktarvy.
Gilead also announced results of a pooled analysis from two Phase III studies showing Biktarvy continued to be highly effective and well-tolerated in treatment-naïve patients age 50 and older over three years of treatment. Importantly, participants experienced no clinically significant differences in key measures such as bone density, renal laboratory markers or weight.
In July, Gilead announced data demonstrating the safety and efficacy of Biktarvy in virologically suppressed adults ages 65 and older, including those with common comorbidities such as diabetes (22 percent), hypertension (55 percent), cardiovascular disease (24 percent), and dyslipidemia, which is an abnormal amount of lipids in the blood (59 percent). At 48 weeks, 92 percent of those who switched to Biktarvy maintained virologic suppression, achieving HIV RNA<50 copies/mL.
Gilead also announced a new data analysis from multiple studies evaluating drug resistance, including the first study to investigate a switch to Biktarvy in virologically suppressed study participants in which some of the participants had a history of treatment failure or suspected pre-existing nucleoside reverse transcriptase inhibitor resistance. The study showed infrequent and similar viral blips (when study participants experienced a temporary viral load at or above 50 copies/mL) among study participants switching to Biktarvy, as compared to the comparator arm. The results support further evaluation of whether the once-daily, single tablet regimen Biktarvy may potentially be an effective and well-tolerated option for adults with a history of treatment failure or pre-existing resistance.
Even with Biktarvy’s rapid uptake in 2019, the drug’s FTC/TAF cousin Genvoya still generated $3.93 billion in sales for the year, a decline of 15 percent. Genvoya’s decline grew a bit steeper in the first half of 2020, with sales down 17.8 percent to $1.64 billion.
Truvada, Gilead’s leading FTC/TDF HIV product, generated $2.81 billion in sales for the company during 2019, a decline of 6.1 percent due to patients switching to newer regimens containing FTC/TAF, partially offset by the increased usage of Truvada for PrEP. In the first half of 2020, sales of Truvada declined another 40.1 percent to $793 million. Teva Pharmaceuticals was expected to launch a generic version of Truvada in late September 2020.
Gilead’s only remaining non-HIV blockbuster in 2019 was sofosbuvir/velpatasvir, also known as Epclusa, for hepatitis C. The combination product generated $1.97 billion in sales for the year, almost identical to 2018. In the first half of 2020, sales declined by 8.6 percent to $899 million. Company leaders blamed this decline on lower patient starts attributable to a decrease in HCP visits and screenings due to the COVID-19 pandemic.
In March, FDA approved a supplemental new drug application for Epclusa for the treatment of people with chronic hepatitis C infection as young as 6 years of age or weighing at least 17 kg, regardless of HCV genotype or liver disease severity. The recommended dosage of Epclusa in children ages 6 years and older is based on weight and liver function. Epclusa is the first pan-genotypic, protease inhibitor-free regimen approved in the United States for adults and children.
About 23,000-46,000 children in the United States are living with HCV. Children born to mothers with HCV are a growing concern, increasing in prevalence by 60 percent from 2011 to 2014. Additionally, engagement in high-risk practices, such as intravenous drug use, is an increasingly common route of HCV transmission in adolescents and young adults.
The approval of Epclusa was based on data from a Phase II, open-label clinical trial (Study 1143) that enrolled 175 children who were treated with Epclusa for 12 weeks, of which 173 were included in the efficacy analysis. In children 12 to <18 years old, treatment with Epclusa resulted in a cure rate (SVR12) of 93 percent in those with genotype 1 HCV infection and 100 percent in those with genotype 2, genotype 3, genotype 4, and genotype 6 HCV infection. In children 6 to <12 years old, the SVR rate was 93 percent in those with genotype 1 HCV infection, 91 percent in those with genotype 3 HCV infection, and 100 percent in those with genotype 2 and genotype 4 HCV infection.
Another FTC/TAF HIV product, Odefsey, brought in $1.66 billion in sales for Gilead in 2019, an improvement of 3.6 percent. Sales of Odefsey edged up by 0.9 percent to $791 million in the first half of 2020.
Gilead’s fourth-leading FTC/TAF HIV product in 2019 was Descovy, with sales of $1.5 billion, down 5.1 percent from the previous year. Descovy made a comeback in the first half of 2020, though, with sales rising 25 percent to $875 million. According to company leaders, this was due to increased use of Descovy for PrEP.
In March, Gilead presented longer-term results from the DISCOVER trial of Descovy for pre-exposure prophylaxis (PrEP), demonstrating continued non-inferior efficacy and continued favorable changes in key markers of renal and bone safety at Week 96 compared with Truvada for PrEP. These results were achieved in the overall study population of men and transgender women at risk for HIV infection, as well as in study sub-populations of participants age 50 and older, those younger than 25 years, and those with moderate renal impairment. A separate analysis of the DISCOVER trial demonstrated that Descovy and Truvada were effective and well-tolerated in Black and Hispanic/Latinx participants.
The 96-week analysis of the DISCOVER trial (Oral 2940) demonstrated significant differences in key markers of bone and renal safety in study participants across different age groups. These differences were also observed in the overall population, in addition to differences in lipid parameters and change in baseline weight. The long-term clinical significance of these differences in renal, bone, and lipid parameters are not known; however, these measures are important to consider as people at risk increasingly use PrEP for longer periods of time.
At Week 96, statistically significant differences in measurements of renal safety favoring Descovy were observed in the overall trial population, as well as in older participants and in those with moderate renal impairment (baseline eGFR=60-≤90 mL/min). In participants older than 50 years of age, those receiving Descovy showed a smaller decrease in median estimated glomerular filtration rate compared with those receiving Truvada (-1 mL/min versus -6 mL/min) at Week 96. Key differences favoring Descovy were also observed in markers of proximal tubular function (β2-microglobulin:creatinine ratio and retinol binding protein:creatinine ratio). Among participants with moderate renal impairment, those randomized to Descovy had smaller changes in eGFR and markers of proximal tubular function. In this sub-group, eGFR increased by 3 mL/min among those taking Descovy and decreased by 1 mL/min in those taking Truvada.
The analysis also found changes in bone mineral density favoring Descovy in the overall trial population and among participants younger than 25 years of age. At Week 96 in participants younger than 25 years, spine BMD increased by 1.39 percent in the Descovy group and decreased by 1.2 percent in the Truvada group. Hip BMD increased 1.21 percent from baseline in the Descovy group and decreased by 1.7 percent in the Truvada group.
Study participants receiving Descovy had stable lipid levels through 96 weeks, whereas those receiving Truvada had decreases in lipid levels after 48 and 96 weeks. Fasting glucose levels were similar between the two groups. Participants in the Truvada group showed smaller mean weight increases than those in the Descovy group (+0.5 kg vs. +1.7 kg at Week 96). These findings are consistent with the lower lipid levels and decreased weight previously observed with TDF.
Gilead’s leading product that does not treat an infectious disease, the oncologic Yescarta, showed impressive signs of growth in the first half of 2020, with sales rising 37 percent to $296 million. According to company leaders, this was primarily due to continued uptake in Europe. Yescarta is presently indicated for the treatment of adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy. In September, Kite submitted a supplemental biologics license application to FDA for Yescarta for the treatment of relapsed or refractory follicular lymphoma and marginal zone lymphoma after two or more prior lines of systemic therapy. Yescarta was previously granted Breakthrough Therapy Designation by FDA for these indications. If approved, Yescarta would become the first chimeric antigen receptor (CAR) T cell therapy approved for the treatment of relapsed or refractory indolent non-Hodgkin lymphoma. The sBLA submission is supported by data from the primary analysis of the Phase II ZUMA-5 trial.
In the pipeline
In March, Gilead announced data from clinical and preclinical studies exploring the use of GS-6207, an investigational, novel, first-in-class inhibitor of HIV-1 capsid function, as a potential long-acting therapy for people living with HIV. Results from a Phase Ib proof-of-concept study of a subcutaneous formulation showed antiviral activity with GS-6207 through the last day of monotherapy, Day 10, with significantly greater reductions in HIV-1 RNA versus placebo across all treatment groups.
Phase I data in healthy volunteers evaluating an oral tablet formulation found GS-6207 to be generally safe and well-tolerated with a pharmacokinetic profile supporting once-a-week administration without regard to food. GS-6207 is an investigational agent that is being developed as a component of a long-acting regimen. GS-6207 disrupts HIV capsid, a multimeric shell that is essential to viral replication, at multiple stages throughout the viral life cycle. FDA granted Breakthrough Therapy Designation for the development of GS-6207 for the treatment of HIV-1 infection in heavily treatment-experienced patients with multi-drug resistance in combination with other antiretroviral drugs.
Also in March, Gilead announced results from a Phase Ib trial evaluating the company’s investigational toll-like receptor 7 agonist vesatolimod as part of a human immunodeficiency virus cure research program. These findings mark the first clinical data showing TLR7 stimulation by vesatolimod is associated with a modestly increased time to viral rebound compared to placebo, as well as enhanced immune function and decreased levels of intact HIV DNA.
This randomized, double-blind, placebo-controlled study evaluated 25 people living with HIV who had demonstrated partial viral suppression (HIV RNA of 50 to 5,000 copies per mL) prior to starting ART, a group referred to as “HIV controllers.” Participants received 10 biweekly doses of the TLR7 agonist vesatolimod or placebo while continuing ART, followed by a treatment interruption in which they stopped therapy and were carefully monitored for viral rebound and safety.
The study found that vesatolimod was associated with a longer period of viral suppression following treatment interruption compared with placebo. The median time to viral rebound (>50 copies/mL) was 4.1 weeks for the vesatolimod group compared with 3.9 weeks for placebo. For rebound to >200 copies/mL, the median time was five weeks for vesatolimod compared with four weeks for placebo. Four individuals in the vesatolimod group had no virologic rebound (>50 c/mL) for six or more weeks.
During May, Gilead and Galapagos NV announced positive topline results from SELECTION, a randomized, double-blind, placebo-controlled, Phase IIb/III trial evaluating the efficacy and safety of the investigational, oral, once-daily, selective JAK1 inhibitor filgotinib in 1,348 biologic-naïve or biologic-experienced adult patients with moderately to severely active ulcerative colitis.
Filgotinib 200 milligrams achieved all primary endpoints in the study, inducing clinical remission at Week 10 and maintaining clinical remission at Week 58 in a significantly higher proportion of patients compared with placebo. Filgotinib 100 milligrams did not achieve statistically significant clinical remission at Week 10.
In this trial, clinical remission was defined as an endoscopic subscore of 0 or 1, rectal bleeding subscore of 0, and ≥ 1 point decrease in stool frequency from baseline to achieve a subscore of 0 or 1. Among the biologic-naïve cohort, 52 percent of patients had a baseline Mayo Clinic Score of nine or higher. In the biologically-experienced cohort, 74 percent of patients had a baseline MCS of nine or higher, and 51 percent were previously treated with two different classes of biologics (TNFα antagonists and an integrin receptor antagonist).
Among biologic-naïve patients, a statistically significant higher proportion of patients achieved clinical remission at Week 10 when treated with filgotinib 200 milligrams (26.1 percent) compared with placebo (15.3 percent). Among biologic-experienced patients, a statistically significant higher proportion of patients achieved clinical remission at Week 10 when treated with filgotinib 200 milligrams (11.5 percent) compared with placebo (4.2 percent).
Patients who achieved clinical response or remission after 10 weeks of treatment with filgotinib 100 milligrams or 200 milligrams were subsequently re-randomized to their induction dose of filgotinib or placebo in a 2:1 ratio and treated through Week 58. Both doses of filgotinib achieved the primary endpoint in this maintenance trial. At Week 58, 37.2 percent of biologic-naïve and biologic-experienced patients receiving filgotinib 200 milligrams achieved clinical remission, compared with 11.2 percent treated with placebo. Of patients receiving filgotinib 100 milligrams, 23.8 percent achieved clinical remission at Week 58, compared with 13.5 percent treated with placebo.
In June, Gilead announced updated results from a single-arm, open-label Phase Ib trial of magrolimab (recently acquired in the Forty Seven transaction) in combination with azacitidine in previously untreated patients with higher-risk myelodysplastic syndrome and previously untreated patients with acute myeloid leukemia who are ineligible for intensive chemotherapy, including patients with TP53-mutant AML, a high unmet need population.
At the time of the data cut-off, 68 patients had been treated with magrolimab plus azacitidine, including 39 patients with previously untreated higher-risk MDS and 29 patients with previously untreated AML. Of 33 MDS patients who were evaluable for efficacy, 91 percent achieved an objective response (response assessments per 2006 IWG MDS criteria) including 42 percent with a complete response. Responses to magrolimab and azacitidine also deepened over time, as the CR rate with at least six months of follow-up was 56 percent in MDS patients.
In AML, 64 percent of patients evaluable for efficacy achieved an objective response (response assessments per 2017 AML ELN criteria), including 56 percent with a CR or a CR with incomplete blood count recovery (CRi). Notably in TP53-mutant AML, a treatment refractory and poor prognosis population, 75 percent achieved a CR or CRi. Median duration of response and median overall survival have not yet been reached in MDS, AML, or TP53-mutant AML, with a median follow-up of 5.8, 9.4, and 8.8 months, respectively.
In July, FDA granted accelerated approval to Kite’s Tecartus (brexucabtagene autoleucel, formerly KTE-X19), the first approved chimeric antigen receptor T cell therapy for the treatment of adult patients with relapsed or refractory mantle cell lymphoma. The approval followed a priority review and FDA Breakthrough Therapy Designation and was based on results of ZUMA-2, a single-arm, open-label study in which 87 percent of patients responded to a single infusion of Tecartus, including 62 percent of patients achieving a complete response.
The approval of Tecartus was supported by data from the ongoing, single arm, open-label ZUMA-2 pivotal trial. The study enrolled 74 adult patients with relapsed or refractory MCL who had previously received anthracycline- or bendamustine-containing chemotherapy, an anti-CD20 antibody therapy and a Bruton tyrosine kinase inhibitor (ibrutinib or acalabrutinib). The primary endpoint was objective response rate per the Lugano Classification (2014), defined as the combined rate of CR and partial responses as assessed by an Independent Radiologic Review Committee.
In the study, 87 percent of patients responded to a single infusion of Tecartus, including 62 percent of patients who achieved a CR. Among all patients, follow-up was at least six months after their first objective disease response. Median duration of response has not yet been reached.
Also in July, Gilead announced data from an ongoing Phase I study which showed that a sustained-delivery subcutaneous formulation of GS-6207, now called lenacapavir, sustained predicted therapeutic concentrations for at least six months following a single 900 milligram dose.
In August, Gilead submitted a new drug application to FDA for Veklury (remdesivir), an investigational antiviral for the treatment of patients with COVID-19. Veklury is available in the United States under an Emergency Use Authorization for the treatment of hospitalized patients with severe COVID-19. The filing is the final tier of the rolling NDA submission that was initiated on April 8, 2020. European regulators had granted a conditional marketing authorization for Veklury in July.
The filing was supported by data from two randomized, open-label, multi-center Phase III clinical studies of Veklury conducted by Gilead and the Phase III randomized, placebo-controlled study of Veklury conducted by the National Institute of Allergy and Infectious Diseases. These studies demonstrated that treatment with Veklury resulted in faster time to recovery compared with placebo and that a 5-day or 10-day treatment duration led to similar clinical improvement.
That same month, FDA expanded the Emergency Use Authorization enabling use of Veklury to treat all hospitalized patients with COVID-19, in addition to the previous authorization for patients hospitalized with severe COVID-19. The expanded EUA was based on results from the Phase III SIMPLE trial evaluating Veklury in hospitalized patients with moderate COVID-19 pneumonia, as well as results from the National Institute of Allergy and Infectious Diseases ACTT-1 trial in hospitalized patients with a range of disease severity.
Results from the Phase III SIMPLE study confirmed top-line results previously announced on June 1, 2020. The primary endpoint evaluated patients at Day 11 on a 7-point ordinal scale and found patients randomized to a 5-day course of Veklury plus standard of care were 65 percent more likely to have an improvement in clinical status compared with those randomized to standard of care alone (OR, 1.65).
For patients in the 10-day Veklury group, the improvement in clinical status at Day 11 was not statistically different compared with the standard of care group (OR, 1.31).
Additionally during August, FDA issued a complete response letter for the NDA for filgotinib for moderately to severely active rheumatoid arthritis. The U.S. regulatory agency requested data from the MANTA and MANTA-RAy studies before completing its review of the NDA. The MANTA and MANTA-RAy studies are designed to assess whether filgotinib has an impact on sperm parameters. FDA additionally expressed concerns regarding the overall benefit/risk profile of the filgotinib 200 milligram dose.
International
Fuel poverty in England is probably 2.5 times higher than government statistics show
The top 40% most energy efficient homes aren’t counted as being in fuel poverty, no matter what their bills or income are.
Share this:



The cap set on how much UK energy suppliers can charge for domestic gas and electricity is set to fall by 15% from April 1 2024. Despite this, prices remain shockingly high. The average household energy bill in 2023 was £2,592 a year, dwarfing the pre-pandemic average of £1,308 in 2019.
The term “fuel poverty” refers to a household’s ability to afford the energy required to maintain adequate warmth and the use of other essential appliances. Quite how it is measured varies from country to country. In England, the government uses what is known as the low income low energy efficiency (Lilee) indicator.
Since energy costs started rising sharply in 2021, UK households’ spending powers have plummeted. It would be reasonable to assume that these increasingly hostile economic conditions have caused fuel poverty rates to rise.
However, according to the Lilee fuel poverty metric, in England there have only been modest changes in fuel poverty incidence year on year. In fact, government statistics show a slight decrease in the nationwide rate, from 13.2% in 2020 to 13.0% in 2023.
Our recent study suggests that these figures are incorrect. We estimate the rate of fuel poverty in England to be around 2.5 times higher than what the government’s statistics show, because the criteria underpinning the Lilee estimation process leaves out a large number of financially vulnerable households which, in reality, are unable to afford and maintain adequate warmth.

Energy security
In 2022, we undertook an in-depth analysis of Lilee fuel poverty in Greater London. First, we combined fuel poverty, housing and employment data to provide an estimate of vulnerable homes which are omitted from Lilee statistics.
We also surveyed 2,886 residents of Greater London about their experiences of fuel poverty during the winter of 2022. We wanted to gauge energy security, which refers to a type of self-reported fuel poverty. Both parts of the study aimed to demonstrate the potential flaws of the Lilee definition.
Introduced in 2019, the Lilee metric considers a household to be “fuel poor” if it meets two criteria. First, after accounting for energy expenses, its income must fall below the poverty line (which is 60% of median income).
Second, the property must have an energy performance certificate (EPC) rating of D–G (the lowest four ratings). The government’s apparent logic for the Lilee metric is to quicken the net-zero transition of the housing sector.
In Sustainable Warmth, the policy paper that defined the Lilee approach, the government says that EPC A–C-rated homes “will not significantly benefit from energy-efficiency measures”. Hence, the focus on fuel poverty in D–G-rated properties.
Generally speaking, EPC A–C-rated homes (those with the highest three ratings) are considered energy efficient, while D–G-rated homes are deemed inefficient. The problem with how Lilee fuel poverty is measured is that the process assumes that EPC A–C-rated homes are too “energy efficient” to be considered fuel poor: the main focus of the fuel poverty assessment is a characteristic of the property, not the occupant’s financial situation.
In other words, by this metric, anyone living in an energy-efficient home cannot be considered to be in fuel poverty, no matter their financial situation. There is an obvious flaw here.
Around 40% of homes in England have an EPC rating of A–C. According to the Lilee definition, none of these homes can or ever will be classed as fuel poor. Even though energy prices are going through the roof, a single-parent household with dependent children whose only income is universal credit (or some other form of benefits) will still not be considered to be living in fuel poverty if their home is rated A-C.
The lack of protection afforded to these households against an extremely volatile energy market is highly concerning.
In our study, we estimate that 4.4% of London’s homes are rated A-C and also financially vulnerable. That is around 171,091 households, which are currently omitted by the Lilee metric but remain highly likely to be unable to afford adequate energy.
In most other European nations, what is known as the 10% indicator is used to gauge fuel poverty. This metric, which was also used in England from the 1990s until the mid 2010s, considers a home to be fuel poor if more than 10% of income is spent on energy. Here, the main focus of the fuel poverty assessment is the occupant’s financial situation, not the property.
Were such alternative fuel poverty metrics to be employed, a significant portion of those 171,091 households in London would almost certainly qualify as fuel poor.
This is confirmed by the findings of our survey. Our data shows that 28.2% of the 2,886 people who responded were “energy insecure”. This includes being unable to afford energy, making involuntary spending trade-offs between food and energy, and falling behind on energy payments.
Worryingly, we found that the rate of energy insecurity in the survey sample is around 2.5 times higher than the official rate of fuel poverty in London (11.5%), as assessed according to the Lilee metric.
It is likely that this figure can be extrapolated for the rest of England. If anything, energy insecurity may be even higher in other regions, given that Londoners tend to have higher-than-average household income.
The UK government is wrongly omitting hundreds of thousands of English households from fuel poverty statistics. Without a more accurate measure, vulnerable households will continue to be overlooked and not get the assistance they desperately need to stay warm.
Torran Semple receives funding from Engineering and Physical Sciences Research Council (EPSRC) grant EP/S023305/1.
John Harvey does not work for, consult, own shares in or receive funding from any company or organisation that would benefit from this article, and has disclosed no relevant affiliations beyond their academic appointment.
european uk pandemicInternational
Looking Back At COVID’s Authoritarian Regimes
After having moved from Canada to the United States, partly to be wealthier and partly to be freer (those two are connected, by the way), I was shocked,…
Share this:


After having moved from Canada to the United States, partly to be wealthier and partly to be freer (those two are connected, by the way), I was shocked, in March 2020, when President Trump and most US governors imposed heavy restrictions on people’s freedom. The purpose, said Trump and his COVID-19 advisers, was to “flatten the curve”: shut down people’s mobility for two weeks so that hospitals could catch up with the expected demand from COVID patients. In her book Silent Invasion, Dr. Deborah Birx, the coordinator of the White House Coronavirus Task Force, admitted that she was scrambling during those two weeks to come up with a reason to extend the lockdowns for much longer. As she put it, “I didn’t have the numbers in front of me yet to make the case for extending it longer, but I had two weeks to get them.” In short, she chose the goal and then tried to find the data to justify the goal. This, by the way, was from someone who, along with her task force colleague Dr. Anthony Fauci, kept talking about the importance of the scientific method. By the end of April 2020, the term “flatten the curve” had all but disappeared from public discussion.
Now that we are four years past that awful time, it makes sense to look back and see whether those heavy restrictions on the lives of people of all ages made sense. I’ll save you the suspense. They didn’t. The damage to the economy was huge. Remember that “the economy” is not a term used to describe a big machine; it’s a shorthand for the trillions of interactions among hundreds of millions of people. The lockdowns and the subsequent federal spending ballooned the budget deficit and consequent federal debt. The effect on children’s learning, not just in school but outside of school, was huge. These effects will be with us for a long time. It’s not as if there wasn’t another way to go. The people who came up with the idea of lockdowns did so on the basis of abstract models that had not been tested. They ignored a model of human behavior, which I’ll call Hayekian, that is tested every day.
These are the opening two paragraphs of my latest Defining Ideas article, “Looking Back at COVID’s Authoritarian Regimes,” Defining Ideas, March 14, 2024.
Another excerpt:
That wasn’t the only uncertainty. My daughter Karen lived in San Francisco and made her living teaching Pilates. San Francisco mayor London Breed shut down all the gyms, and so there went my daughter’s business. (The good news was that she quickly got online and shifted many of her clients to virtual Pilates. But that’s another story.) We tried to see her every six weeks or so, whether that meant our driving up to San Fran or her driving down to Monterey. But were we allowed to drive to see her? In that first month and a half, we simply didn’t know.
Read the whole thing, which is longer than usual.
(0 COMMENTS) budget deficit coronavirus covid-19 white house fauci trump canadaInternational
Problems After COVID-19 Vaccination More Prevalent Among Naturally Immune: Study
Problems After COVID-19 Vaccination More Prevalent Among Naturally Immune: Study
Authored by Zachary Stieber via The Epoch Times (emphasis…
Share this:

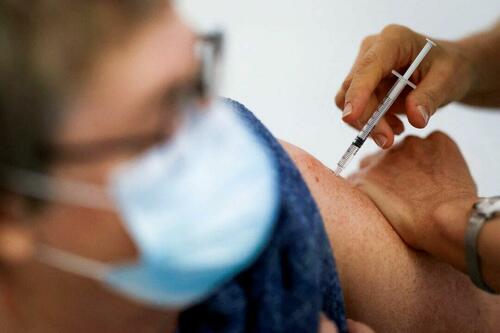
Authored by Zachary Stieber via The Epoch Times (emphasis ours),
People who recovered from COVID-19 and received a COVID-19 shot were more likely to suffer adverse reactions, researchers in Europe are reporting.
{kind=link}
Participants in the study were more likely to experience an adverse reaction after vaccination regardless of the type of shot, with one exception, the researchers found.
Across all vaccine brands, people with prior COVID-19 were 2.6 times as likely after dose one to suffer an adverse reaction, according to the new study. Such people are commonly known as having a type of protection known as natural immunity after recovery.
People with previous COVID-19 were also 1.25 times as likely after dose 2 to experience an adverse reaction.
The findings held true across all vaccine types following dose one.
Of the female participants who received the Pfizer-BioNTech vaccine, for instance, 82 percent who had COVID-19 previously experienced an adverse reaction after their first dose, compared to 59 percent of females who did not have prior COVID-19.
The only exception to the trend was among males who received a second AstraZeneca dose. The percentage of males who suffered an adverse reaction was higher, 33 percent to 24 percent, among those without a COVID-19 history.
“Participants who had a prior SARS-CoV-2 infection (confirmed with a positive test) experienced at least one adverse reaction more often after the 1st dose compared to participants who did not have prior COVID-19. This pattern was observed in both men and women and across vaccine brands,” Florence van Hunsel, an epidemiologist with the Netherlands Pharmacovigilance Centre Lareb, and her co-authors wrote.
There were only slightly higher odds of the naturally immune suffering an adverse reaction following receipt of a Pfizer or Moderna booster, the researchers also found.
The researchers performed what’s known as a cohort event monitoring study, following 29,387 participants as they received at least one dose of a COVID-19 vaccine. The participants live in a European country such as Belgium, France, or Slovakia.
Overall, three-quarters of the participants reported at least one adverse reaction, although some were minor such as injection site pain.
Adverse reactions described as serious were reported by 0.24 percent of people who received a first or second dose and 0.26 percent for people who received a booster. Different examples of serious reactions were not listed in the study.
Participants were only specifically asked to record a range of minor adverse reactions (ADRs). They could provide details of other reactions in free text form.
“The unsolicited events were manually assessed and coded, and the seriousness was classified based on international criteria,” researchers said.
The free text answers were not provided by researchers in the paper.
“The authors note, ‘In this manuscript, the focus was not on serious ADRs and adverse events of special interest.’” Yet, in their highlights section they state, “The percentage of serious ADRs in the study is low for 1st and 2nd vaccination and booster.”
Dr. Joel Wallskog, co-chair of the group React19, which advocates for people who were injured by vaccines, told The Epoch Times: “It is intellectually dishonest to set out to study minor adverse events after COVID-19 vaccination then make conclusions about the frequency of serious adverse events. They also fail to provide the free text data.” He added that the paper showed “yet another study that is in my opinion, deficient by design.”
Ms. Hunsel did not respond to a request for comment.
She and other researchers listed limitations in the paper, including how they did not provide data broken down by country.
The paper was published by the journal Vaccine on March 6.
The study was funded by the European Medicines Agency and the Dutch government.
No authors declared conflicts of interest.
Some previous papers have also found that people with prior COVID-19 infection had more adverse events following COVID-19 vaccination, including a 2021 paper from French researchers. A U.S. study identified prior COVID-19 as a predictor of the severity of side effects.
Some other studies have determined COVID-19 vaccines confer little or no benefit to people with a history of infection, including those who had received a primary series.
The U.S. Centers for Disease Control and Prevention still recommends people who recovered from COVID-19 receive a COVID-19 vaccine, although a number of other health authorities have stopped recommending the shot for people who have prior COVID-19.
Another New Study
In another new paper, South Korean researchers outlined how they found people were more likely to report certain adverse reactions after COVID-19 vaccination than after receipt of another vaccine.
The reporting of myocarditis, a form of heart inflammation, or pericarditis, a related condition, was nearly 20 times as high among children as the reporting odds following receipt of all other vaccines, the researchers found.
The reporting odds were also much higher for multisystem inflammatory syndrome or Kawasaki disease among adolescent COVID-19 recipients.
Researchers analyzed reports made to VigiBase, which is run by the World Health Organization.
“Based on our results, close monitoring for these rare but serious inflammatory reactions after COVID-19 vaccination among adolescents until definitive causal relationship can be established,” the researchers wrote.
The study was published by the Journal of Korean Medical Science in its March edition.
Limitations include VigiBase receiving reports of problems, with some reports going unconfirmed.
Funding came from the South Korean government. One author reported receiving grants from pharmaceutical companies, including Pfizer.


Net Zero, The Digital Panopticon, & The Future Of Food


Problems After COVID-19 Vaccination More Prevalent Among Naturally Immune: Study


For-profit nursing homes are cutting corners on safety and draining resources with financial shenanigans − especially at midsize chains that dodge public scrutiny


Looking Back At COVID’s Authoritarian Regimes


Trump nearly derailed democracy once − here’s what to watch out for in reelection campaign


‘Excess Mortality Skyrocketed’: Tucker Carlson and Dr. Pierre Kory Unpack ‘Criminal’ COVID Response


Health Officials: Man Dies From Bubonic Plague In New Mexico


MIPIM 2024 Reflects Mixed Feelings on CRE Recovery


Riley Gaines Explains How Women’s Sports Are Rigged To Promote The Trans Agenda


Five Aerospace Investments to Buy as Wars Worsen Copy
-

 Uncategorized3 weeks ago
Uncategorized3 weeks agoAll Of The Elements Are In Place For An Economic Crisis Of Staggering Proportions
-

 International1 week ago
International1 week agoEyePoint poaches medical chief from Apellis; Sandoz CFO, longtime BioNTech exec to retire
-

 Uncategorized4 weeks ago
Uncategorized4 weeks agoCalifornia Counties Could Be Forced To Pay $300 Million To Cover COVID-Era Program
-

 Uncategorized3 weeks ago
Uncategorized3 weeks agoApparel Retailer Express Moving Toward Bankruptcy
-

 Uncategorized4 weeks ago
Uncategorized4 weeks agoIndustrial Production Decreased 0.1% in January
-

 International7 days ago
International7 days agoWalmart launches clever answer to Target’s new membership program
-

 Spread & Containment2 days ago
Spread & Containment2 days agoIFM’s Hat Trick and Reflections On Option-To-Buy M&A
-

 Uncategorized4 weeks ago
Uncategorized4 weeks agoRFK Jr: The Wuhan Cover-Up & The Rise Of The Biowarfare-Industrial Complex