International
Company of the Year 2020: Bristol-Myers Squibb — Patience Rewarded
Company of the Year 2020: Bristol-Myers Squibb — Patience Rewarded
Share this:


Five years after the company’s portfolio bottomed out, Bristol-Myers Squibb markets three of the world’s six best-selling pharma brands.
By Joshua Slatko • josh.slatko@medadnews.com
Bristol-Myers Squibb Co.
430 E. 29th Street, 14th Floor
New York, NY 10016
Phone: 212-546-4000
Website: bms.com
FINANCIAL PERFORMANCE
(All figures are in millions of dollars, except EPS)
2019
Revenue $26,145
Net income $3,460
Diluted EPS $2.01
R&D expense $6,148
1H 2020
Revenue $20,910
Net income $(846)
Diluted EPS $(0.38)
R&D expense $4,894
BEST-SELLING Rx PRODUCTS
(All sales are in millions of dollars)
2019
Eliquis $7,929
Opdivo $7,204
Orencia $2,977
Sprycel $2,110
Yervoy $1,489
Revlimid $1,299*
Baraclude $555
* – Sales with BMS after completion of the Celgene acquisition in November 2019.
1H 2020
Revlimid $5,799
Eliquis $4,804
Opdivo $3,419
Orencia $1,464
Pomalyst/Imnovid $1,458
Sprycel $1,032
Yervoy $765
Abraxane $608
Vidaza $284
Outcomes Creativity Index Score: 0
Manny Awards – N/A
Cannes Lions – N/A
LIA: Health & Wellness – N/A
Clio Health – N/A
One Show: HW&P – N/A
MM&M Awards – N/A
Global Awards – N/A
Creative Floor Awards – N/A
When Giovanni Caforio, MD, took over as CEO of Bristol-Myers Squibb in May 2015, the company’s annual top-line revenue had just fallen below $16 billion and its leading product, the $2 billion Abilify, had gone generic the month before. But the seeds of future success had already been planted. Opdivo earned the drug’s first approval from FDA in December 2014, sales of Eliquis had just more than quadrupled to $774 million, and BMS was starting to look like it might really be what the leadership team had been starting to call it: an Immuno-Oncology company. Having risen through the company’s oncology divisions, Dr. Caforio was the man for the moment. Five years later, Opdivo is the world’s third-best-selling oncologic, Eliquis is the world’s leading cardiovascular brand, and BMS’ acquisition of Celgene – a move that would have been barely conceivable back in 2015 – has brought the $10 billion oncology brand Revlimid into the fold as well, alongside Pomalyst (tracking towards $3 billion in annual sales), the recently approved products Reblozyl, Zeposia and Onureg, and the high-potential developmental CAR T compounds liso-cel and ide-cel, among others. For this extraordinary turnaround, for the courageous and ultimately successful all-in bets on Opdivo and Eliquis, and for the aggressive pursuit and capture of Celgene to firm up both the top line and the pipeline, Med Ad News is pleased to name Bristol-Myers Squibb as Company of the Year.

“By all measures, 2019 was a transformative year for Bristol-Myers Squibb as we progressed our strategy through the acquisition of Celgene, delivered strong operational and financial performance, and continued to drive important science for patients,” said Giovanni Caforio, MD, CEO of Bristol-Myers Squibb.
“By all measures, 2019 was a transformative year for Bristol-Myers Squibb as we progressed our strategy through the acquisition of Celgene, delivered strong operational and financial performance, and continued to drive important science for patients,” Dr. Caforio said at the end of 2019. “With an expanded portfolio of high-performing brands, eight potential commercial launch opportunities, a deep and broad early pipeline, and the financial flexibility to continue to invest in innovation, the company enters 2020 uniquely positioned to transform patients’ lives through science and create long-term sustainable growth.”
Helped along by a month and change of legacy Celgene revenue, BMS’ top line in 2019 was $26.15 billion, an improvement of 15.9 percent compared with the previous year. Due in part to the impact of amortization of acquired intangible assets and other costs from the Celgene transaction, though, net income for the year declined 30.1 percent to $3.46 billion and diluted earnings per share were down a dollar to $2.01. In the first half of 2020, the first full half of legacy Celgene, top-line sales were up by 71.5 percent to $20.91 billion. However, due to more amortization of acquired intangible assets – $4.67 billion of it – and other Celgene-related costs, net income fell into the red at negative $846 million and EPS was -$0.38. BMS executives are estimating that full-year EPS for 2020 will fall between -$0.06 and $0.09; without all the one-time Celgene charges and costs, though, they are projecting full-year 2020 EPS at between $6.10 and $6.25.
Acquisitions & partnerships
In January, BMS and Nektar Therapeutics announced a new joint development plan to advance bempegaldesleukin (bempeg) plus Opdivo into multiple new registrational trials.
The revision to the companies’ strategic collaboration agreement includes a new joint development plan under which Nektar and Bristol-Myers Squibb will expand the active clinical development program for bempeg plus nivolumab from three ongoing registrational trials in first-line metastatic melanoma, first-line cisplatin-ineligible metastatic urothelial cancer and first-line metastatic renal cell carcinoma (RCC) to include two additional registrational trials in adjuvant melanoma and in muscle-invasive bladder cancer. In addition, a Phase I/II dose escalation and expansion study will be initiated to evaluate bempeg plus nivolumab in combination with axitinib in first-line RCC in order to support a future registrational trial. The costs for these studies will be shared based upon the cost-sharing outlined in the terms of the original collaboration agreement. Also as part of the new strategic collaboration agreement, Bristol-Myers Squibb will independently conduct and fund a Phase I/II dose optimization and expansion study in first-line non-small-cell lung cancer with bempeg and nivolumab.
In February, BMS and BioMotiv, a mission-driven drug development accelerator associated with The Harrington Project for discovery and development, that advances breakthrough discoveries from research institutions into therapeutics, announced the launch of Anteros Pharmaceuticals, a biotechnology company focused on developing a new class of drugs for fibrotic and other inflammatory diseases. The intellectual property behind Anteros was first developed by Yale University and in-licensed by Bristol-Myers Squibb and subsequently assigned to Anteros. This is the first company BioMotiv and Bristol-Myers Squibb have launched since executing their Strategic Partnership Agreement, as previously announced in September 2019.
Under the terms of the partnership, Bristol-Myers Squibb is contributing the IP, data, and reagents for a series of small molecules against an undisclosed mechanism, and BioMotiv, through the formation of Anteros Pharmaceuticals, working in close partnership with Yale, is solely responsible for research and development. Once Anteros nominates a pre-clinical candidate, Bristol-Myers Squibb has the option to acquire the company from BioMotiv under pre-agreed terms.
During March, Bristol-Myers Squib and Voluntis announced a collaboration agreement to create and investigate digital therapeutic solutions that will support cancer patients. Taking advantage of Theraxium Oncology, Voluntis’ core platform for digital therapeutics in oncology, the collaboration is evaluating potential solutions that will support management of patient symptoms and remote monitoring by healthcare providers.
According to company leaders, the goal is that the digital therapeutic, once researched and developed, would provide patients access to a mobile app that would support treatment and track symptoms. The app will be developed to embed evidence-based algorithms intended to provide patients with real-time recommendations for self-management of symptoms related to their therapy. The parties will also investigate how the solution could enable patients to more effectively communicate with their health care providers, capture and track symptoms, and receive a personalized supportive care plan.
In August 2020, BMS and Forbius, a privately held, clinical-stage protein engineering company that designs and develops biotherapeutics for the treatment of cancer and fibrotic diseases, announced that they had entered into a definitive agreement under which Bristol-Myers Squibb would acquire Forbius. The acquisition was completed during September.
Forbius has developed a portfolio of highly selective and potent inhibitors of TGF-beta 1 and 3, which are key mediators of immunosuppression and fibrosis. The transaction included an upfront payment and future success-based milestone payments. Prior to closing, Forbius’ non-TGF-beta assets were transferred to a newly formed private company, which is retained by Forbius’ existing shareholders.
Under this transaction, Bristol-Myers Squibb acquired Forbius’ TGF-beta program, including the program’s lead investigational asset, AVID200. TGF-beta is a key cytokine that regulates various cell processes, including regulation of the immune system. Selective inhibition of TGF-beta 1 and 3 may enhance anti-tumor efficacy by acting synergistically with immunotherapy. AVID200 is undergoing Phase 1 development for oncology and fibrosis.
Also in August, BMS and Dragonfly Therapeutics Inc. announced that they had entered into a definitive agreement under which Bristol-Myers Squibb would be granted the global exclusive license to Dragonfly’s interleukin-12 (IL-12) investigational immunotherapy program, including its extended half-life cytokine DF6002. DF6002 is a monovalent IL-12 immunoglobulin Fc fusion protein proposed to achieve strong anti-tumor efficacy by establishing an inflammatory tumor microenvironment necessary for productive anti-tumor responses.
Under the agreement, Bristol-Myers Squibb is responsible for the development and any subsequent commercialization of DF6002 and its related products worldwide, including strategic decisions, regulatory responsibilities, funding, and manufacturing. Dragonfly will receive $475 million in near-term upfronts, and is eligible to receive performance-based development, regulatory and commercial milestone payments. In addition, Dragonfly will receive up to 24 percent royalties on worldwide net sales.

The Opdivo development train continued to roll during 2020, with two additional FDA approvals in NSCLC and another in HCC.
Dragonfly received FDA clearance in May 2020 for its investigational new drug application to develop DF6002. In addition, Dragonfly has an ongoing Phase I/II clinical trial for patients with advanced solid tumors, which began in July 2020. BMS intends to advance the research and development of DF6002 in oncology and hematology.
Opdivo
Sales of the oncologic Opdivo rose 7 percent in 2019 to $7.2 billion. While international sales were up by 15 percent due to higher demand as a result of approvals for additional indications in 2018 and launches in Europe and Asia, sales growth in the United States was 2 percent, primarily due to a smaller previously treated advanced lung cancer market and increased competition for the Opdivo+Yervoy combination in kidney cancer. Company leaders expect this trend to continue until the market stabilizes or new indications are approved and launched. In the first half of 2020 sales of Opdivo declined by 5.7 percent to $3.42 billion. According to company leaders, this was again due to the smaller previously treated advanced lung cancer market, as well as lower demand due to COVID-19, primarily lower new patient starts and patient visits.
In February, BMS announced five-year follow-up results from the Phase III CheckMate -025 study, which continue to demonstrate that treatment with Opdivo delivers superior overall survival (OS) rate and objective response rate (ORR) in patients with previously treated advanced or metastatic renal cell carcinoma compared to those treated with everolimus.
With an extended minimum follow-up of 64 months, patients treated with Opdivo continued to demonstrate OS benefit with 26 percent of patients alive compared to 18 percent of patients treated with everolimus. Additionally, the percentage of patients experiencing an objective response was 23 percent for Opdivo versus 4 percent for everolimus and the median duration of response for Opdivo was also maintained longer than for everolimus (18.2 months versus 14 months, respectively).
Also in February, BMS announced updated results from the Phase III CheckMate -214 study evaluating the combination of Opdivo plus Yervoy versus sunitinib in patients with previously untreated advanced or metastatic renal cell carcinoma. With a minimum follow-up of 42 months, the combination of Opdivo plus Yervoy continued to show superior OS, ORR, duration of response, and complete response rates.

Sales of Yervoy in the first half of 2020 rose by 1.9 percent to $765 million.
A significant OS benefit was observed in both patients from the intermediate- and poor-risk and the intent-to-treat populations treated with Opdivo plus Yervoy compared to those treated with sunitinib alone. Of the patients treated with Opdivo plus Yervoy who experienced a complete response, per independent review, that response was ongoing in 84 percent and 86 percent of patients in the IP and ITT populations, respectively.
More than half of advanced RCC patients treated with the Opdivo plus Yervoy combination were alive after four years across the entire study population of the Phase 3 CheckMate -214 trial, as reported by Bristol-Myers Squibb in September 2020. With the longest follow-up for an immunotherapy-based combination in previously untreated advanced RCC, Opdivo plus Yervoy continued to demonstrate superior, long-term OS and durable responses versus sunitinib. These sustained benefits were observed across the primary patient population, those with intermediate- and poor-risk prognostic factors, and in the intention-to-treat (ITT, i.e. all randomized) patient population.
During March, FDA approved Opdivo 1 mg/kg plus Yervoy 3 mg/kg (injections for intravenous use) to treat hepatocellular carcinoma (HCC) in patients who have been previously treated with sorafenib. Approval for this indication was granted under accelerated approval based on overall response rate and duration of response seen in the Opdivo + Yervoy cohort of the Phase I/II CheckMate -040 trial. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials. Opdivo + Yervoy is the only dual immunotherapy approved by FDA in this setting. The therapy features a potentially synergistic mechanism of action that targets two different checkpoints (PD-1 and CTLA-4) and works in complementary ways.
In the CheckMate -040 cohort of HCC patients previously treated with sorafenib, after a minimum follow up of 28 months, 33 percent of patients responded to treatment with Opdivo + Yervoy; 8 percent had a complete response, and 24 percent had a partial response. Duration of responses ranged from 4.6 to 30.5-plus months, with 88 percent lasting at least six months, 56 percent at least 12 months, and 31 percent at least 24 months. Overall response rate and DOR were assessed by Blinded Independent Central Review using Response Evaluation Criteria in Solid Tumors version 1.1. ORR assessed by BICR using modified RECIST was 35 percent, with a CR reported in 12 percent of patients and a PR reported in 22 percent of patients.
In April, BMS and Exelixis Inc. announced that CheckMate -9ER, a pivotal Phase III trial evaluating Opdivo in combination with Cabometyx compared to sunitinib in previously untreated advanced or metastatic renal cell carcinoma, met its primary endpoint of progression-free survival (PFS) at final analysis, as well as the secondary endpoints of OS at a pre-specified interim analysis, and ORR.
CheckMate -9ER results were reported in September in which Opdivo in combination with Cabometyx showed superior OS and doubled median PFS and ORR with a favorable safety profile compared to sunitinib.
In May, FDA approved Opdivo 360 mg plus Yervoy 1 mg/kg (injections for intravenous use) given with two cycles of platinum-doublet chemotherapy for the first-line treatment of adult patients with metastatic or recurrent non-small cell lung cancer with no EGFR or ALK genomic tumor aberrations. The therapy was approved for patients with squamous or non-squamous disease and regardless of PD-L1 expression. This application was reviewed under FDA’s Real-Time Oncology Review pilot program, which aims to ensure that safe and effective treatments are available to patients as early as possible.
This approval for Opdivo + Yervoy with limited chemotherapy was based on the pre-specified interim analysis from the Phase III CheckMate -9LA trial in which Opdivo + Yervoy combined with two cycles of platinum-doublet chemotherapy demonstrated superior OS versus chemotherapy regardless of PD-L1 expression or tumor histology (minimum 8.1 months follow up). Median OS was 14.1 months versus 10.7 months, respectively. In a follow-up analysis at 12.7 months, the hazard ratio improved numerically to 0.66, with mOS of 15.6 months and 10.9 months. At one year, 63 percent of patients treated with Opdivo + Yervoy with limited chemotherapy and 47 percent of those treated with chemotherapy were still alive.
Also in May, FDA approved Opdivo 3 mg/kg plus Yervoy 1 mg/kg (injections for intravenous use) for the first-line treatment of adults with metastatic non-small cell lung cancer whose tumors express PD-L1 (≥1 percent) as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations.
This approval was based on Part 1a of the Phase III CheckMate -227 trial in which Opdivo + Yervoy demonstrated superior OS versus chemotherapy regardless of tumor histology with a minimum follow up of 29.3 months. The median OS was 17.1 months versus 14.9 months. In the trial, 63 percent of patients treated with Opdivo + Yervoy and 56 percent treated with chemotherapy were alive at one year, and 40 percent and 33 percent at two years, respectively. At three years (median 43.1 months follow up), 33 percent of patients treated with Opdivo + Yervoy and 22 percent of those treated with chemotherapy were still alive. As assessed by Blinded Independent Central Review, the confirmed overall response rate with a minimum follow up of 28.3 months was 36 percent with Opdivo + Yervoy (5.8 percent complete response and 30.1 percent partial response) and 30 percent with chemotherapy (1.8 percent CR and 28.2 percent PR). Among patients who responded, the median duration of response was 23.2 months for patients treated with Opdivo + Yervoy and 6.2 months for chemotherapy.
In June, FDA approved Opdivo for the treatment of patients with unresectable advanced, recurrent, or metastatic esophageal squamous cell carcinoma after prior fluoropyrimidine- and platinum-based chemotherapy. This application was granted Priority Review Designation by FDA, and the approval was based on the Phase III ATTRACTION-3 trial in which Opdivo demonstrated superior OS versus taxane chemotherapy (investigator’s choice of docetaxel or paclitaxel). The median OS was 10.9 months for Opdivo compared to 8.4 months for docetaxel or paclitaxel. Opdivo is the first approved immunotherapy in this setting regardless of tumor PD-L1 expression level.
In August 2020, BMS announced that the Phase III CheckMate -577 trial evaluating Opdivo as an adjuvant therapy for patients with resected esophageal or gastroesophageal junction (GEJ) cancer met its primary endpoint of disease-free survival at a pre-specified interim analysis. In the trial, treatment with Opdivo following neoadjuvant chemoradiation therapy and complete surgical resection demonstrated a statistically significant improvement in the primary endpoint of disease-free survival (DFS) compared to placebo in the all-randomized population. The safety profile of Opdivo was consistent with previously reported studies. This is the second tumor, in addition to melanoma, where Opdivo has demonstrated a benefit in the adjuvant setting.
Bristol-Myers Squibb announced results in September from the Phase 3 CheckMate -577 trial in which adjuvant treatment with Opdivo demonstrated a statistically significant and clinically meaningful improvement in DFS compared to placebo in patients with esophageal or GEJ cancer following neoadjuvant chemoradiation therapy and tumor resection.
BMS announced during August that CheckMate -649, a pivotal Phase III trial evaluating Opdivo plus chemotherapy compared to chemotherapy alone as a first-line treatment for metastatic gastric cancer, GEJ cancer or esophageal adenocarcinoma, met both primary endpoints of OS at a pre-specified interim analysis and progression-free survival at final analysis in patients whose tumors express PD-L1 with a combined positive score ≥ 5. The OS benefit was also observed in the all-randomized population. Opdivo is the first PD-1 inhibitor to demonstrate superior OS and PFS in combination with chemotherapy when compared to chemotherapy alone in patients with gastric cancer, GEJ, cancer or esophageal adenocarcinoma.
Bristol Myers Squibb reported in September primary results from CheckMate -649 in which first-line treatment with Opdivo plus chemotherapy demonstrated a statistically significant and clinically meaningful improvement in OS and PFS of patients with unresectable advanced or metastatic gastric cancer, GEJ cancer or esophageal adenocarcinoma compared to treatment with chemotherapy alone.
In early October 2020, Opdivo 360 mg every three weeks plus Yervoy 1 mg/kg every six weeks was approved by the U.S. FDA for the first-line treatment of adult patients with unresectable malignant pleural mesothelioma (MPM). The U.S. approval is based on a pre-specified interim analysis from the Phase 3 CheckMate -743 trial in which Opdivo + Yervoy (n=303) demonstrated superior overall survival versus the platinum-based standard of care chemotherapy, with a median OS of 18.1 months (95 percent CI: 16.8 to 21.5) versus 14.1 months (95 percent CI: 12.5 to 16.2), respectively. These results were observed after 22.1 months of minimum follow-up, according to Bristol-Myers Squibb. At two years, 41 percent of patients treated with Opdivo + Yervoy were alive and 27 percent with chemotherapy.

Eliquis took over as the world’s top-selling cardiovascular medicine in 2019 with sales of $7.93 billion.
Opdivo + Yervoy is the first new systemic therapy in more than 15 years to be approved by the FDA in this setting. The marketing approval marks the third indication for Opdivo + Yervoy-based treatments in thoracic cancers and seventh indication overall.
Other product performances
The cardiovascular drug Eliquis took over as BMS’ leading brand by revenue in 2019 with growth of 23.2 percent and sales of $7.93 billion for the year, passing Xarelto to become the world’s No. 1 cardiovascular brand. According to BMS executives, U.S. sales of Eliquis increased due to higher demand, partially offset by higher Medicare Part D coverage gap cost share, while international sales rose due to higher demand. In the first half of 2020 sales of Eliquis rose another 21.1 percent to $4.8 billion. Eliquis is indicated for the prevention of stroke in adults with nonvalvular atrial fibrillation, and the prevention and treatment of venous thromboembolism disorders.
The autoimmune product Orencia generated sales of $2.98 billion for BMS in 2019, an improvement of 9.9 percent. According to company leaders, this was driven by higher demand and higher average net selling price. Sales of Orencia in the first half of 2020 edged up by 3.2 percent to $1.46 billion, slowed by the impact of the COVID-19 pandemic. Orencia is indicated for adult patients with moderate to severe active rheumatoid arthritis and psoriatic arthritis and is also indicated for reducing signs and symptoms in certain pediatric patients with moderately to severely active polyarticular juvenile idiopathic arthritis.
In June, BMS announced results from the open-label switch period of Early AMPLE, a Phase IV exploratory biomarker study assessing the differences by which Orencia and another treatment, adalimumab, interfere with disease progression in moderate-to-severe early rheumatoid arthritis patients who tested positive (seropositive) for certain autoantibodies. Findings of the open-label switch period showed that early seropositive RA patients treated with Orencia demonstrated substantial clinical improvements at week 48, sustaining the level of responses achieved at week 24 compared to adalimumab. In seropositive patients switching from adalimumab to Orencia, the efficacy responses generally increased over the open-label period to week 48.
The efficacy responses observed at 24 weeks with Orencia were sustained at week 48 in the patients who continued on Orencia. At week 48, ACR 20/50/70 responses with Orencia in the non-switch arm were 78, 63, and 50, respectively. At week 24, ACR 20/50/70 responses with Orencia were 83, 73, and 50, respectively; ACR 20/50/70 scores for adalimumab at week 24 were 63, 45, and 30, respectively.
In the patients who switched from adalimumab to Orencia, while the trial was not powered to show superiority or non-inferiority, the efficacy responses generally increased over the open-label period through week 48. ACR 20/50/70 scores for patients who switched from adalimumab to Orencia were 75, 63, and 38, respectively, at week 48.
Overall, patients with a well-known genetic marker of RA prognosis, called the “Shared Epitope,” who continued on Orencia achieved numerically higher responses than the broader seropositive patient population at week 48, indicating the potential importance of SE as a predictor of response to Orencia. ACR 20/50/70 responses were 77, 67 and 53, respectively, for SE+ patients continuing on Orencia.
Sales of the leukemia drug Sprycel rose by 5.5 percent in 2019 to $2.11 billion. Company leaders said this was due to higher average net selling price and higher demand in the United States. In the first half of 2020, Sprycel sales rose 2.9 percent to $1.03 billion, with demand declining in the United States and generic competition internationally. Sprycel is indicated for the first-line treatment of adults with Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase and the treatment of adults with chronic, accelerated, or myeloid or lymphoid blast phase CML with resistance or intolerance to prior therapy.
The oncologic Yervoy generated $1.49 billion in sales for BMS in 2019, an improvement of 12 percent. According to company executives, this was due to higher demand and higher average net selling price in the United States and approvals for additional indications and launches primarily in Europe and Japan in 2018 internationally. Sales of Yervoy in the first half of 2020 rose by 1.9 percent to $765 million, held back by increased competition for the Opdivo + Yervoy combination for kidney cancer.

After joining the BMS portfolio via the Celgene acquisition, the oncologic Revlimid is trending towards more than $11 billion in revenue in 2020.
While they did not have time to add much to BMS’ 2019 numbers since the Celgene deal closed in November, two legacy Celgene products are going to have a major impact on BMS’ top line in 2020: Revlimid and Pomalyst/Imnovid. Revlimid, the world’s leading oncology drug by revenue in 2018, generated $10.82 billion in sales and growth of 11.7 percent between Celgene and BMS in 2019, losing its oncology crown only because Merck’s Keytruda grew faster. In the first half of 2020 sales of Revlimid were $5.8 billion, up 9.2 percent compared with the medicine’s first-half 2019 amount while still under the Celgene umbrella. The multiple myeloma drug Pomalyst generated sales of more than $2.16 billion in 2019 – the numbers are a bit unclear due to the timing of the Celgene acquisition – and brought in another $1.46 billion in the first half of 2020, up 24 percent compared with the drug’s first-half 2019 performance at Celgene. Revlimid is indicated in combination with dexamethasone for the treatment of patients with multiple myeloma and as a single agent as a maintenance therapy in patients with multiple myeloma following autologous hematopoietic stem cell transplant. Pomalyst is indicated for patients with multiple myeloma who have received at least two prior therapies including lenalidomide and a proteasome inhibitor and have demonstrated disease progression on or within 60 days of completion of the last therapy.
In May, FDA approved Pomalyst for patients with AIDS-related Kaposi sarcoma whose disease has become resistant to highly active antiretroviral therapy (HAART), or in patients with Kaposi sarcoma who are HIV-negative. Pomalyst was granted accelerated approval, Breakthrough Therapy designation and Orphan Drug designation in these indications based on overall response rates observed in a Phase I/II open label, single-arm clinical trial (12-C-0047). Continued approval may be contingent upon verification and description of clinical benefit in a confirmatory trial. Pomalyst is the first new treatment option available for those with Kaposi sarcoma in more than 20 years.

The legacy Celgene oncologic Pomalyst enjoyed sales growth of nearly a quarter in the first half of 2020 and earned a new indication from FDA for AIDS-related Kaposi sarcoma.
The approval of Pomalyst was based on the findings of a Phase I/II open-label, single-arm study conducted evaluating the safety, pharmacokinetics and efficacy of Pomalyst in patients with HIV-positive and HIV-negative symptomatic Kaposi sarcoma, the majority of whom had advanced disease. A total of 28 patients (18 HIV-positive, 10 HIV-negative) received 5 milligrams of Pomalyst, once daily for 21 of 28-day cycles, until disease progression or unacceptable toxicity. All HIV-positive patients continued concomitant HAART. The trial excluded patients with symptomatic pulmonary or visceral Kaposi sarcoma, history of venous or arterial thromboembolism, or procoagulant disorders. Patients received thromboprophylaxis with aspirin 81 milligrams once daily throughout therapy. The median time to first response was 1.8 months.
The primary endpoint of the study was overall response rate, which included complete response, clinical complete response, and partial response, as assessed by investigators according to the AIDS Clinical Trial Group Oncology Committee response criteria for Kaposi sarcoma. For all patients, the ORR was 71 percent with 14 percent of patients achieving CR and 57 percent of patients achieving a PR, respectively. The median duration of response for all patients was 12.1 months. Additionally, half (50 percent) of patients who responded maintained a response at more than 12 months with Pomalyst.
In the pipeline
FDA in February 2020 accepted for Priority Review BMS’ biologics license application for lisocabtagene maraleucel (liso-cel), the company’s autologous anti-CD19 chimeric antigen receptor T-cell immunotherapy with a defined composition of purified CD8+ and CD4+ CAR T cells for the treatment of adult patients with relapsed or refractory (R/R) large B-cell lymphoma after at least two prior therapies. A similar application was subsequently accepted for review by European regulators in July.
The BLA, submitted by Juno Therapeutics, a wholly owned subsidiary of BMS, was based on the safety and efficacy results from the TRANSCEND NHL 001 trial, evaluating liso-cel in 268 patients with R/R large B-cell lymphoma, including diffuse large B-cell lymphoma, high-grade lymphoma, primary mediastinal B-cell lymphoma, and Grade 3B follicular lymphoma. TRANSCEND NHL 001 is the largest study of CD19-directed CAR T cells to support a BLA to date.
In March, FDA approved Zeposia (ozanimod) 0.92 mg for the treatment of adults with relapsing forms of multiple sclerosis (RMS), including clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease. Zeposia, an oral medication taken once daily, is the only approved sphingosine-1-phosphate receptor modulator that offers RMS patients an initiation with no genetic test and no label-based first-dose observation required for patients. The product was subsequently approved by the European Commission in May, and launched the following month.
The approval is based on data from the largest pivotal, head-to-head RMS studies with an active comparator to date: the randomized, active-controlled Phase III SUNBEAM (safety and efficacy of Zeposia versus interferon beta-1a in relapsing multiple sclerosis) and RADIANCE (safety and efficacy of the selective sphingosine 1-phosphate receptor modulator Zeposia in relapsing multiple sclerosis) Part B clinical trials of more than 2,600 adults. In both trials – as compared to Avonex (interferon beta-1a), Zeposia delivered powerful efficacy as measured by annualized relapse rat, as well as on the number and size of brain lesions. Zeposia demonstrated a relative reduction in ARR versus Avonex of 48 percent through one year and 38 percent at two years.
Bristol Myers Squibb reported in September interim results from the Phase 3 open-label extension study DAYBREAK, showing the long-term efficacy and safety profile of Zeposia in patients with RMS. The study included 2,494 patients who had previously completed a Phase 1, 2 or 3 Zeposia clinical trial and who had an average treatment time of 35.4 months while in DAYBREAK.
In March, BMS announced topline results from ELOQUENT-1, a Phase III, randomized, open-label trial evaluating the combination of Empliciti plus Revlimid and dexamethasone (ERd), versus Revlimid and dexamethasone alone (Rd), in patients with newly diagnosed, previously untreated multiple myeloma who are transplant ineligible. Both treatments were administered continuously until disease progression. At final analysis, the addition of Empliciti did not show a statistically significant improvement in progression-free survival, the study’s primary endpoint.
In April, FDA approved Reblozyl (luspatercept-aamt), the first erythroid maturation agent, for the treatment of anemia failing an erythropoiesis stimulating agent and requiring 2 or more red blood cell units over 8 weeks in adult patients with very low- to intermediate-risk myelodysplastic syndromes with ring sideroblasts or with myelodysplastic/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis.
The approval of Reblozyl was based on the findings of MEDALIST, a Phase III, randomized, double blind, placebo-controlled, multi-center study evaluating the efficacy and safety of Reblozyl in patients with IPSS-R-defined very low-, low- and intermediate-risk non-del(5q) myelodysplastic syndromes (MDS) with ring sideroblasts. All patients were red blood cell transfusion-dependent and were either refractory or intolerant to prior erythropoiesis-stimulating agent therapy or were ESA naïve and unlikely to respond due to endogenous serum erythropoietin ≥200 U/L, and had no prior treatment with disease modifying agents.
In the trial, a significantly greater proportion of patients receiving Reblozyl achieved independence from RBC transfusions for at least eight weeks during the first 24 weeks of the trial compared with those receiving placebo, meeting the study’s primary endpoint. Additionally, a significantly greater proportion of patients receiving Reblozyl vs. placebo achieved at least 12 weeks of independence from transfusions within the first 24 and 48 weeks of the study.
In June, BMS announced results from True North, a pivotal Phase III trial evaluating oral Zeposia as an induction and maintenance therapy for adult patients with moderate to severe ulcerative colitis. True North met both primary endpoints, demonstrating highly statistically significant results for induction of clinical remission at Week 10 and in maintenance at Week 52. The study also met key secondary endpoints of clinical response and endoscopic improvement in induction at Week 10 and in maintenance at Week 52. Bristol-Myers Squibb is also investigating Zeposia for the treatment of moderately to severely active Crohn’s disease in the ongoing Phase III YELLOWSTONE clinical trial program.
Also in June, the European Commission approved Reblozyl for the treatment of adult patients with transfusion-dependent anemia due to very low-, low-, and intermediate-risk myelodysplastic syndromes with ring sideroblasts, who had an unsatisfactory response or are ineligible for erythropoietin-based therapy, and for adult patients with transfusion-dependent anemia associated with beta thalassemia. Reblozyl is the first erythroid maturation agent approved in the European Union, representing a new class of therapy for eligible patients. This approval was based on data from the pivotal Phase III MEDALIST and BELIEVE studies, evaluating the ability of Reblozyl to effectively address anemia associated with MDS and beta thalassemia, respectively.
BELIEVE is a Phase III, randomized, double-blind, placebo-controlled multi-center study comparing Reblozyl plus BSC versus placebo plus BSC in adults who require regular RBC transfusions (6-20 RBC units per 24 weeks with no transfusion-free period greater than 35 days during that period) due to beta thalassemia.
The trial showed a statistically significant improvement in RBC transfusion burden during weeks 13 to 24 compared to the baseline 12-week interval prior to randomization (21.4 percent Reblozyl versus 4.5 percent placebo), meeting the study’s primary endpoint. The trial also met the secondary endpoint of transfusion burden reduction of at least 33 percent (with a reduction of at least two units) during weeks 37 to 48, which was achieved in a significantly greater proportion of patients receiving Reblozyl versus placebo. The trial also met an exploratory endpoint, with 70.5 percent of patients treated with Reblozyl achieving at least a 33 percent reduction in RBC transfusion burden of at least two units for any 12 consecutive weeks compared to the 12-week interval prior to treatment, compared to 29.5 percent of patients on placebo.
In July 2020, BMS and partner developer bluebird bio Inc. announced the submission of a biologics license application (BLA) to FDA for idecabtagene vicleucel (ide-cel; bb2121), the companies’ investigational B-cell maturation antigen (BCMA)-directed chimeric antigen receptor (CAR) T cell immunotherapy, for the treatment of adult patients with relapsed and refractory multiple myeloma. This submission provides further details on the Chemistry, Manufacturing, and Controls (CMC) module to address the outstanding regulatory requests from FDA in May 2020 following the original BLA submission from March 2020.
The submission is based on results from the pivotal Phase II KarMMa study evaluating the efficacy and safety of ide-cel in relapsed and refractory multiple myeloma patients exposed to an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 antibody. Ide-cel has been granted Breakthrough Therapy Designation by FDA, and PRIority MEdicines (PRIME) designation and validation of its marketing authorization application by the European Medicines Agency for relapsed and refractory multiple myeloma.
The BLA for idecabtagene vicleucel was accepted for FDA Priority Review in September. Ide-cel represents the first CAR T cell therapy accepted for regulatory review for multiple myeloma. The U.S. regulatory agency set a Prescription Drug User Fee Act goal date of March 27, 2021.
In August 2020, BMS announced that the Phase III IDHENTIFY study evaluating Idhifa (enasidenib) plus best supportive care versus conventional care regimens, which include best supportive care only, azacitidine plus BSC, low-dose cytarabine plus BSC, or intermediate-dose cytarabine plus BSC, did not meet the primary endpoint of overall survival in patients with relapsed or refractory acute myeloid leukemia with an isocitrate dehydrogenase-2 (IDH2) mutation.
Bristol-Myers Squibb received full approval in the United States for Idhifa for the treatment of adult patients with R/R AML with an IDH2 mutation as detected by a U.S. Food and Drug Administration (FDA)-approved test in August 2017. Idhifa is the first FDA-approved therapy for patients with R/R AML and positive for an IDH2 mutation, which represents up to 19 percent of AML patients. Idhifa is also approved in Australia and Canada.
In September 2020, FDA approved Onureg (azacitidine 300 milligram tablets, CC-486) for the continued treatment of adult patients with acute myeloid leukemia who achieved first complete remission (CR) or CR with incomplete blood count recovery (CRi) following intensive induction chemotherapy and who are not able to complete intensive curative therapy. AML is one of the most common acute leukemias in adults.
The approval was based on results from the pivotal Phase III QUAZAR AML-001 study in which treatment with Onureg resulted in a statistically significant and clinically meaningful improvement in overall survival, the study’s primary endpoint, of nearly 10 months compared to placebo. Median OS from time of randomization was greater than two years (24.7 months) among patients who received Onureg compared to 14.8 months among patients receiving placebo. Onureg was continued until disease progression or unacceptable toxicity.
International
TikTok Ban Obscures Chinese Stock Gold Rush
No one wants to invest in China right now. The country’s stock market is teetering on the brink of collapse. And it is about to lose its biggest foothold…
Share this:


No one wants to invest in China right now.
The country’s stock market is teetering on the brink of collapse.
And it is about to lose its biggest foothold in America — TikTok.
Yet, beneath its crumbling economy, military weather balloons and blatant propaganda tools lie some epic opportunities…
…if you have the stomach and the knowledge.
Because as Jim Woods wrote in his newsletter last month:
“China has been so battered for so long, that there is a lot of deep value here for the ‘blood in the ‘’red’’ streets’ investors.”
And boy was he right.
However, this battle-tested veteran didn’t recommend buying individual Chinese stocks.
He was more interested in the exchange-traded funds (ETFs) like the CHIQ.
And here’s why…
Predictable Manipulation
China’s heavy-handed approach creates gaping economic inefficiencies.
When markets falter, President Xi calls on his “national team” to prop up prices.
$17 billion flowed into index-tracking funds in January as the Hang Sang fell over 13% while the CSI dropped over 7%.
Jim Woods saw this coming from a mile away.
In late February, he highlighted the Chinese ETF CHIQ in late February, which has rallied rather nicely since then.
This ETF focuses on the Chinese consumer, a recent passion project for the central government.
You see, around 2018, when President Xi decided to smother his own economy, notable shifts were already taking place.
The once burgeoning retail market had slowed markedly. Developers left cities abandoned, including weird copies of Paris (Tianducheng) and England.
Source: Shutterstock
So, Xi and co. shifted the focus to the consumer… which went terribly.
For starters, a lot of the consumer wealth was tied up in real estate.
Then you had a growing population of unemployed younger adults who didn’t have any money to spend.
Once the pandemic hit, everything collapsed.
That’s why it took China far longer to recover even a sliver of its former economy.
While it’s not the growth engine of the early 2000s, the old girl still has some life left in it.
As Jim pointed out, China’s consumer spending rebounded nicely in Q4 2023.

Source: National Bureau of Statistics of China
Combined with looser central bank policy, it was only a matter of time before Chinese stocks caught a lift.
The resurgence may be largely tied to China’s desire to travel. After all, its people have been cooped up longer than any other country.
But make no mistake, this doesn’t make China a long-term investment.
Beyond what most people understand about China’s politics, there’s a little-known fact about how they treat foreign investors.
Money in. Nothing out.
When we buy a stock, we’re taking partial ownership in that company. This entitles us to a portion of the profits (or assets).
That doesn’t happen with Chinese companies.
American depository receipts (ADRs) aren’t actual shares of a company. It’s a note that the intermediary ties to shares of the company they own overseas.
So, we can only own Chinese companies indirectly.
But there’s another key feature you probably weren’t aware of.
Many of the Chinese companies we, as Americans invest in, don’t pay dividends. In fact, a much smaller percentage of Chinese companies pay any dividends.
Alibaba is a perfect example.
Despite generating billions of dollars in cash every year, it doesn’t pay dividends.
What do its managers do with the money?
Other than squirreling away $80 billion on its balance sheets, they do share buybacks.
Plenty of investors will tell you that’s even better than dividends.
But you have no legal ownership rights in China. So, what is that ADR in reality?
We’d argue nothing but paper profits at best, and air at worst.
That’s why it’s flat-out dangerous to own shares of individual Chinese companies long-term.
Any one of them can be nationalized at any moment.
Chinese ETFs reduce that risk through diversification, similar to junk bond funds.
Short of an all-out ban, like between the United States and Russia, the majority of the ETF holdings should remain intact.
Opportunistic Investing
If China is so unstable, and capable of changing at a moment’s notice, how can investors uncover pockets of value?
As Jim showed with his ETF selection, you can have some sector or thematic idea so long as you have the data to support it.
China, like any large institution, isn’t going to change its broad economic policies overnight.
As long as you study the general movements of the government, you can steer clear of the catastrophic zones and towards the diamond caves.
Because when things look THIS bad, you know the opportunities are even juicier.
But rather than try to run this maze solo, take this opportunity to check out Jim Woods’ latest report on China.
In it, he details the broad economic themes driving the Chinese government, and how to exploit them for gain.
Click here to explore Jim Woods’ report.
The post TikTok Ban Obscures Chinese Stock Gold Rush appeared first on Stock Investor.
stocks pandemic real estate etf consumer spending gold russia chinaInternational
The Great Escape… of UK Unemployment Reporting
https://bondvigilantes.com/wp-content/uploads/2024/03/1-the-great-escape-of-uk-unemployment-reporting-1024×576.pngThe Bank of England Monetary Policy Committee…
Share this:

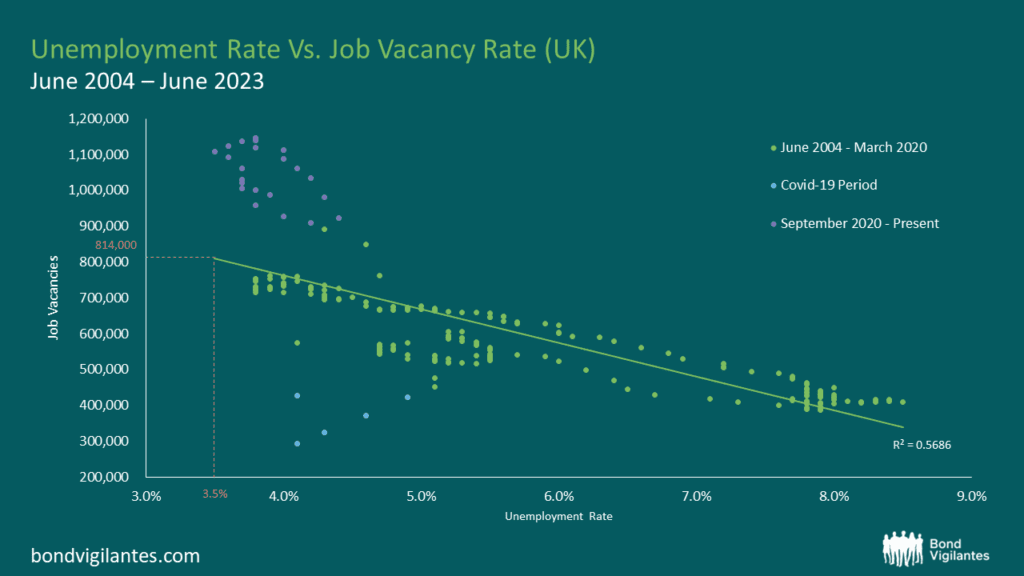
The Bank of England Monetary Policy Committee potentially has a problem: it requires data to make its labour market forecasts and assessments, but the unemployment statistics have become increasingly unreliable. This is because the Labour Force Survey participation rate (on which the unemployment figures are based) has fallen below 50% since 2018 and has been as low as 15% recently[1]. What is the solution to this difficult measurement problem? An answer can be found in the classic war film, The Great Escape.
In 1943, the Escape Committee of Stalag Luft III was tasked with digging a tunnel to freedom. Unfortunately, they had a problem. They needed to measure the distance between one of the prisoner’s huts and the forest beyond the prison perimeter, but they had no reliable tools to measure this critical variable. Fortunately they had two mathematicians within the group who came up with a method to gauge the distance to the forest so that the tunnel would be long enough to ensure escape without detection. The idea was to eyeball the distance using a 20 foot tree for scale (the tree was the one ‘accurate’ measurement around which they could work with). They got individual prisoners to gauge the distance from the hut to the tree and then averaged all of the estimates. The critical distance measure was therefore the average of a large sample size of guesstimates. Fortunately, it more or less worked. Happily, modern economists have an equivalent to rely on in the area of unemployment. Their version of the Stalag Luft III tree strategy is something called the Beveridge Curve.
The Beveridge Curve is simply an observed relationship between an economy’s unemployment rate and its job vacancy rate at the same point in time. An excellent exposition can be found in the Bond Vigilantes archive[2]. When you plot the two variables against one another over a given period, the data points disclose a curve. This curve shows us that when unemployment increases, job vacancies decrease and vice versa. I have plotted the current curve below using the available data from the Office for National Statistics (ONS)[3]. The bottom left quadrant of the graph (the blue dots) relate to the Covid-19 era and the top left quadrant (the purple dots) represent the last 2 years’ worth of data. The green dots represent the remaining data from July 2004 to June 2023.
Source: Office for National Statistics, Dataset JP9Z & UNEM

Source: Office for National Statistics, Dataset JP9Z & UNEM
From these charts and new data from the ONS, we can observe that in the UK, the level of unemployment is increasing and that the job vacancy rate is decreasing. At face value, this suggests that current Bank of England monetary policy is working and that the inflation rate is slowing as the economy cools. One could argue that we are on track for a reasonably soft landing. Nothing new so far.
Things become more interesting when we consider the Beveridge Curve in conjunction with the most recent job vacancy data. We are told that there are now 814,000 job vacancies as of the 31st December 2023[4]. Ordinarily, we would use the curve and clearly be able to extrapolate from the Job Vacancy data what our Unemployment figure might be. However, we also know that the current unemployment data is unreliable, which makes this harder. Using our model inclusive of data oddities, we could extrapolate that with 814,000 job vacancies, we might expect an unemployment rate of around 3.5%. Yet, we know that our unemployment figures are unreliable so the question therefore is, how big an increase in unemployment are we likely to see given what we know about job vacancies?
In order to estimate the magnitude of the rise in unemployment, we need to look further afield. If we study the levels of economic inactivity in the UK, we can observe that they have remained stationary at 22%[5] for the last decade. We can also see that the population of the UK has risen over the same period by around 5.91%[6]. Further, we know that the Labour Force Survey (LFS) samples 40,000 households per quarter to obtain its data, but of late has had a response rate of only 15% (6,000 households). Therefore a critical question for policy makers is what is happening with the 85%, the non-responders?
Given the small sample size, it is entirely possible that the LFS suffered survey bias that is being erroneously weighted away. In other words, the LFS compensates for the paucity of response data by accessing other regional population statistics as a legitimate part of their methodology. The problems of non-responders are being addressed in upcoming LFS releases but for the time being, the data is not as clear as it ought to be. With such a small sample size, it seems possible – indeed probable – that unemployment levels are being underreported. This would explain why the current unemployment rate of 3.8%[7] is dramatically lower than the historic average of 6.7% (1971-2023). We see further evidence for this in the forecasts of the UK’s unemployment rate on Bloomberg which have been consistently above the actual levels for the last few published data points. So whilst the published headline figures might be looking reasonable, the underlying story looks like it could be hiding something more sinister.
Through it all, the Beveridge Curve remains a reasonable template. Job vacancies are definitely falling, so we should expect to see unemployment rising. Like the Stalag Luft III measurement solution, the Beveridge Curve offers a constructive way out of our present statistical dilemma. That being said, analogies can only be taken so far. Unfortunately for the inmates of Stalag Luft III, the calculation didn’t quite work and the tunnel came up short. No one actually made a Great Escape. What does this mean for UK unemployment data? Time may tell.
[1] The UK’s ‘official’ labour data is becoming a nonsense (harvard.edu)
[2] https://bondvigilantes.com/blog/2013/11/a-shifting-beveridge-curve-does-the-us-have-a-long-term-structural-unemployment-problem/
[3] Unemployment – Office for National Statistics (ons.gov.uk)
[4] https://www.ons.gov.uk/employmentandlabourmarket/peopleinwork/employmentandemployeetypes/timeseries/jp9z/unem
[5] https://www.ethnicity-facts-figures.service.gov.uk/work-pay-and-benefits/unemployment-and-economic-inactivity/economic-inactivity/latest/#:~:text=data%20shows%20that%3A-,22%25%20of%20working%20age%20people%20in%20England%2C%20Scotland%20and%20Wales,for%20a%20job)%20in%202022
[6] https://www.ons.gov.uk/peoplepopulationandcommunity/populationandmigration/populationestimates/bulletins/annualmidyearpopulationestimates/mid2021
[7] https://www.ons.gov.uk/employmentandlabourmarket/peoplenotinwork/unemployment
unemployment covid-19 monetary policy unemployment ukInternational
Germany Is Running Out Of Money And Debt Levels Are Exploding, Finance Minister Warns
Germany Is Running Out Of Money And Debt Levels Are Exploding, Finance Minister Warns
By John Cody of Remix News
German Finance Minister…
Share this:

{kind=link}

By John Cody of Remix News
German Finance Minister Christian Lindner is warning his own government that state finances are quickly growing out of hand, and the government needs to change course and implement austerity measures. However, the dispute over spending is only expected to escalate, with budget shortfalls causing open clashes among the three-way left-liberal coalition running the country.
{kind=link}
With negotiations kicking off for the 2025 budget, much is at stake. However, the picture has been complicated after the country’s top court ruled that the government could not shift €60 billion in money earmarked for the coronavirus crisis to other areas of the budget, with the court noting that the move was unconstitutional.
Since then, the government has been in crisis mode, and sought to cut the budget in a number of areas, including against the country’s farmers. Those cuts already sparked mass protests, showcasing how delicate the situation remains for the government.

Lindner, whose party has taken a beating in the polls, is desperate to create some distance from his coalition partners and save his party from electoral disaster. The finance minster says the financial picture facing Germany is dire, and that the budget shortfall will only grow in the coming years if measures are not taken to rein in spending.
“In an unfavorable scenario, the increasing financing deficits lead to an increase in debt in relation to economic output to around 345 percent in the long term,” reads the Sustainability Report released by his office. “In a favorable scenario, the rate will rise to around 140 percent of gross domestic product by 2070.”
Under EU law, Germany has limited its debt levels to 60 percent of economic output, which requires dramatic savings. A huge factor is Germany’s rapidly aging population, with a debt explosion on the horizon as more and more citizens head into retirement while tax revenues shrink and the social welfare system grows — in part due to the country’s exploding immigrant population.
Lindner’s partners, the Greens and Social Democrats (SPD), are loath to cut spending further, as this will harm their electoral chances. In fact, Labor Minister Hubertus Heil is pushing for a new pension package that will add billions to the country’s debt, which remarkably, Lindner also supports.
Continue reading at rmx.news


Mistakes Were Made


Home buyers must now navigate higher mortgage rates and prices


Gen Z, The Most Pessimistic Generation In History, May Decide The Election


“Extreme Events”: US Cancer Deaths Spiked In 2021 And 2022 In “Large Excess Over Trend”


Germany Is Running Out Of Money And Debt Levels Are Exploding, Finance Minister Warns


You can strike gold and silver investment opportunities at Costco


TikTok Ban Obscures Chinese Stock Gold Rush


The Great Escape… of UK Unemployment Reporting
-

 Uncategorized4 weeks ago
Uncategorized4 weeks agoAll Of The Elements Are In Place For An Economic Crisis Of Staggering Proportions
-

 International1 week ago
International1 week agoEyePoint poaches medical chief from Apellis; Sandoz CFO, longtime BioNTech exec to retire
-

 Spread & Containment5 days ago
Spread & Containment5 days agoIFM’s Hat Trick and Reflections On Option-To-Buy M&A
-

 Uncategorized1 month ago
Uncategorized1 month agoCalifornia Counties Could Be Forced To Pay $300 Million To Cover COVID-Era Program
-

 Uncategorized3 weeks ago
Uncategorized3 weeks agoApparel Retailer Express Moving Toward Bankruptcy
-

 Uncategorized1 month ago
Uncategorized1 month agoIndustrial Production Decreased 0.1% in January
-

 International1 week ago
International1 week agoWalmart launches clever answer to Target’s new membership program
-

 Uncategorized1 month ago
Uncategorized1 month agoRFK Jr: The Wuhan Cover-Up & The Rise Of The Biowarfare-Industrial Complex