Government
Preview the Cars and Trucks at This Year’s New York Auto Show
After two years of pandemic-related cancellations, the New York Auto Show returns for a 10-day event.
Share this:


After two years of pandemic-related cancellations, the New York Auto Show returns for a 10-day event.
After two years of pandemic-related cancellations, the New York Auto Show returns for a 10-day event that celebrates the latest and greatest the automotive industry has to offer.
With the electric car market expected to more than quadruple from $208.95 billion in 2021 to $957.42 billion, it is not surprise that EVs took up a major part of the conversation at the show.
The cars themselves, as well as everything from vehicle charging stations to battery types, took up nearly an entire level of the Javits Center convention hall.
Governor Kathy Hochul said in a speech the state is encouraging New Yorkers to consider switching to electric at the opening press day on April 13.
"I am proud to welcome the New York International Auto Show back to New York in person this year and I encourage everyone to visit the Auto Show to see how EVs are fast becoming integral to New York's clean energy economy," Hochul said.
TheStreet was able to get an early peek at some of the displays and interactive experiences.
Here are some of the coolest models that car lovers can expect to see in the coming years.
Kia EV9 Concept:
South Korean carmaker Kia (KIMTF) presented a number of models at the auto show and, among them, is the Kia EV9 Concept.
The brand's flagship SUV has a 336-mile range and a battery that can replenish 62 miles in just six minutes. The giant wheels and shiny aquamarine color made it a popular stop at the show.
As many of the cars featured at the show won't be available for purchase for quite some time, Kia's commitment to bring it to buyers in the second half of 2023 was also exciting.
TheStreet
Lamborghini Huracan Tecnica
As Lamborghini moves to electrify its car lineup, cars with only an internal combustion engine will slowly be phased out.
But before that, the luxury carmaker unveiled the Huracan Tecnica. The 5.2-litre V10 has 631 horsepower and can go to 62 miles per hour in 3.2 seconds.
The model will be available for sale this summer but, at a starting cost of $279,630, has a price tag befitting a luxury sports car.

TheStreet
New Chevrolet Silverado EV
General Motors (GM) - Get General Motors Company Report's Chevrolet recently came out with the 2024 Chevrolet Silverado EV.
The brand's first electric pick-up truck model, the car will come in three models: A Work Truck (WT), an off-road Trail Boss, and a loaded Rally Sport Truck.
With the electric truck market now including Ford (F) - Get Ford Motor Company Report F-150 Lightning and the Rivian (RIVN) - Get Rivian Automotive, Inc. Class A Report R1T, Chevrolet hopes to draw customers with its speed, the carmaker promises 60 miles per hour in under 4.5 seconds, and strength, 664 horsepower and a 400-mile range.
The company also recently partnered with Goldman Sachs for a credit card that builds points that can go toward buying, leasing or servicing a GM car.

TheStreet
EV Test Track
One of the most popular attractions was the ability to test-drive the cars themselves, for those who are still not convinced that EVs are up to par with anything gas-powered.
The Chevrolet Bolt EV, Kia EV6 (KIMTF) , Nissan Leaf (NSANF) , VinFast VF8 and Volvo XC40 Recharge Twin (VOLAF) are all available for test-driving on the lower level of the Javits Center.
The track is built to feel like a "drive through the country" and allows drivers to test speeding up, stopping and, of course, making sharp turns.

Airflow Graphite Concept
While car connoisseurs often rail at people discussing a car's color over its features, Chrysler's Airflow Graphite Concept stood out.
It was contrasted with the white electric Chrysler Airflow Concept unveiled at the Consumer Electronics Show for its "sinister yet sophisticated" black look accented with high-gloss copper, glass roof and 22-inch wheels.
As the brand's first all-electric model, it is expected to go on sale by 2025.

TheStreet
Elvis Presley's 1956 Continental Mark II
Fans of car history will be impressed by the 1956 Continental Mark II once owned by Elvis Presley himself.
Normally stored at the Graceland museum in Memphis, the historic model was on display on a turning pedestal at the Lincoln (LINC) - Get Lincoln Educational Services Corporation Report station at the show.
Glistening and full of features like rear suspension and power steering, it could have easily been mistaken for a car from this century.

TheStreet
Government
The War Between Knowledge And Stupidity
The War Between Knowledge And Stupidity
Authored by Bert Olivier via The Brownstone Institute,
Bernard Stiegler was, until his premature…
Share this:


Authored by Bert Olivier via The Brownstone Institute,
Bernard Stiegler was, until his premature death, probably the most important philosopher of technology of the present. His work on technology has shown us that, far from being exclusively a danger to human existence, it is a pharmakon – a poison as well as a cure – and that, as long as we approach technology as a means to ‘critical intensification,’ it could assist us in promoting the causes of enlightenment and freedom.
It is no exaggeration to say that making believable information and credible analysis available to citizens at present is probably indispensable for resisting the behemoth of lies and betrayal confronting us. This has never been more necessary than it is today, given that we face what is probably the greatest crisis in the history of humanity, with nothing less than our freedom, let alone our lives, at stake.
To be able to secure this freedom against the inhuman forces threatening to shackle it today, one could do no better than to take heed of what Stiegler argues in States of Shock: Stupidity and Knowledge in the 21st Century (2015). Considering what he writes here it is hard to believe that it was not written today (p. 15):
The impression that humanity has fallen under the domination of unreason or madness [déraison] overwhelms our spirit, confronted as we are with systemic collapses, major technological accidents, medical or pharmaceutical scandals, shocking revelations, the unleashing of the drives, and acts of madness of every kind and in every social milieu – not to mention the extreme misery and poverty that now afflict citizens and neighbours both near and far.
While these words are certainly as applicable to our current situation as it was almost 10 years ago, Stiegler was in fact engaged in an interpretive analysis of the role of banks and other institutions – aided and abetted by certain academics – in the establishment of what he terms a ‘literally suicidal financial system’ (p. 1). (Anyone who doubts this can merely view the award-winning documentary film of 2010, Inside Job, by Charles Ferguson, which Stiegler also mentions on p.1.) He explains further as follows (p. 2):
Western universities are in the grip of a deep malaise, and a number of them have found themselves, through some of their faculty, giving consent to – and sometimes considerably compromised by – the implementation of a financial system that, with the establishment of hyper-consumerist, drive-based and ‘addictogenic’ society, leads to economic and political ruin on a global scale. If this has occurred, it is because their goals, their organizations and their means have been put entirely at the service of the destruction of sovereignty. That is, they have been placed in the service of the destruction of sovereignty as conceived by the philosophers of what we call the Enlightenment…
In short, Stiegler was writing about the way in which the world was being prepared, across the board – including the highest levels of education – for what has become far more conspicuous since the advent of the so-called ‘pandemic’ in 2020, namely an all-out attempt to cause the collapse of civilisation as we knew it, at all levels, with the thinly disguised goal in mind of installing a neo-fascist, technocratic, global regime which would exercise power through AI-controlled regimes of obedience. The latter would centre on ubiquitous facial recognition technology, digital identification, and CBDCs (which would replace money in the usual sense).
Given the fact that all of this is happening around us, albeit in a disguised fashion, it is astonishing that relatively few people are conscious of the unfolding catastrophe, let alone being critically engaged in disclosing it to others who still inhabit the land where ignorance is bliss. Not that this is easy. Some of my relatives are still resistant to the idea that the ‘democratic carpet’ is about to be pulled from under their feet. Is this merely a matter of ‘stupidity?’ Stiegler writes about stupidity (p.33):
…knowledge cannot be separated from stupidity. But in my view: (1) this is a pharmacological situation; (2) stupidity is the law of the pharmakon; and (3) the pharmakon is the law of knowledge, and hence a pharmacology for our age must think the pharmakon that I am also calling, today, the shadow.
In my previous post I wrote about the media as pharmaka (plural of pharmakon), showing how, on the one hand, there are (mainstream) media which function as ‘poison,’ while on the other there are (alternative) media that play the role of ‘cure.’ Here, by linking the pharmakon with stupidity, Stiegler alerts one to the (metaphorically speaking) ‘pharmacological’ situation, that knowledge is inseparable from stupidity: where there is knowledge, the possibility of stupidity always asserts itself, and vice versa. Or in terms of what he calls ‘the shadow,’ knowledge always casts a shadow, that of stupidity.
Anyone who doubts this may only cast their glance at those ‘stupid’ people who still believe that the Covid ‘vaccines’ are ‘safe and effective,’ or that wearing a mask would protect them against infection by ‘the virus.’ Or, more currently, think of those – the vast majority in America – who routinely fall for the Biden administration’s (lack of an) explanation of its reasons for allowing thousands of people to cross the southern – and more recently also the northern – border. Several alternative sources of news and analysis have lifted the veil on this, revealing that the influx is not only a way of destabilising the fabric of society, but possibly a preparation for civil war in the United States.
There is a different way of explaining this widespread ‘stupidity,’ of course – one that I have used before to explain why most philosophers have failed humanity miserably, by failing to notice the unfolding attempt at a global coup d’etat, or at least, assuming that they did notice it, to speak up against it. These ‘philosophers’ include all the other members of the philosophy department where I work, with the honourable exception of the departmental assistant, who is, to her credit, wide awake to what has been occurring in the world. They also include someone who used to be among my philosophical heroes, to wit, Slavoj Žižek, who fell for the hoax hook, line, and sinker.
In brief, this explanation of philosophers’ stupidity – and by extension that of other people – is twofold. First there is ‘repression’ in the psychoanalytic sense of the term (explained at length in both the papers linked in the previous paragraph), and secondly there is something I did not elaborate on in those papers, namely what is known as ‘cognitive dissonance.’ The latter phenomenon manifests itself in the unease that people exhibit when they are confronted by information and arguments that are not commensurate, or conflict, with what they believe, or which explicitly challenge those beliefs. The usual response is to find standard, or mainstream-approved responses to this disruptive information, brush it under the carpet, and life goes on as usual.
‘Cognitive dissonance’ is actually related to something more fundamental, which is not mentioned in the usual psychological accounts of this unsettling experience. Not many psychologists deign to adduce repression in their explanation of disruptive psychological conditions or problems encountered by their clients these days, and yet it is as relevant as when Freud first employed the concept to account for phenomena such as hysteria or neurosis, recognising, however, that it plays a role in normal psychology too. What is repression?
In The Language of Psychoanalysis (p. 390), Jean Laplanche and Jean-Bertrand Pontalis describe ‘repression’ as follows:
Strictly speaking, an operation whereby the subject attempts to repel, or to confine to the unconscious, representations (thoughts, images, memories) which are bound to an instinct. Repression occurs when to satisfy an instinct – though likely to be pleasurable in itself – would incur the risk of provoking unpleasure because of other requirements.
…It may be looked upon as a universal mental process to so far as it lies at the root of the constitution of the unconscious as a domain separate from the rest of the psyche.
In the case of the majority of philosophers, referred to earlier, who have studiously avoided engaging critically with others on the subject of the (non-)‘pandemic’ and related matters, it is more than likely that repression occurred to satisfy the instinct of self-preservation, regarded by Freud as being equally fundamental as the sexual instinct. Here, the representations (linked to self-preservation) that are confined to the unconscious through repression are those of death and suffering associated with the coronavirus that supposedly causes Covid-19, which are repressed because of being intolerable. The repression of (the satisfaction of) an instinct, mentioned in the second sentence of the first quoted paragraph, above, obviously applies to the sexual instinct, which is subject to certain societal prohibitions. Cognitive dissonance is therefore symptomatic of repression, which is primary.
Returning to Stiegler’s thesis concerning stupidity, it is noteworthy that the manifestations of such inanity are not merely noticeable among the upper echelons of society; worse – there seems to be, by and large, a correlation between those in the upper classes, with college degrees, and stupidity.
In other words, it is not related to intelligence per se. This is apparent, not only in light of the initially surprising phenomenon pertaining to philosophers’ failure to speak up in the face of the evidence, that humanity is under attack, discussed above in terms of repression.
Dr Reiner Fuellmich, one of the first individuals to realise that this was the case, and subsequently brought together a large group of international lawyers and scientists to testify in the ‘court of public opinion’ (see 29 min. 30 sec. into the video) on various aspects of the currently perpetrated ‘crime against humanity,’ has drawn attention to the difference between the taxi drivers he talks to about the globalists’ brazen attempt to enslave humanity, and his learned legal colleagues as far as awareness of this ongoing attempt is concerned. In contrast with the former, who are wide awake in this respect, the latter – ostensibly more intellectually qualified and ‘informed’ – individuals are blissfully unaware that their freedom is slipping away by the day, probably because of cognitive dissonance, and behind that, repression of this scarcely digestible truth.
This is stupidity, or the ‘shadow’ of knowledge, which is recognisable in the sustained effort by those afflicted with it, when confronted with the shocking truth of what is occurring worldwide, to ‘rationalise’ their denial by repeating spurious assurances issued by agencies such as the CDC, that the Covid ‘vaccines’ are ‘safe and effective,’ and that this is backed up by ‘the science.’
Here a lesson from discourse theory is called for. Whether one refers to natural science or to social science in the context of some particular scientific claim – for example, Einstein’s familiar theory of special relativity (e=mc2) under the umbrella of the former, or David Riesman’s sociological theory of ‘inner-’ as opposed to ‘other-directedness’ in social science – one never talks about ‘the science,’ and for good reason. Science is science. The moment one appeals to ‘the science,’ a discourse theorist would smell the proverbial rat.
Why? Because the definite article, ‘the,’ singles out a specific, probably dubious, version of science compared to science as such, which does not need being elevated to special status. In fact, when this is done through the use of ‘the,’ you can bet your bottom dollar it is no longer science in the humble, hard-working, ‘belonging-to-every-person’ sense. If one’s sceptical antennae do not immediately start buzzing when one of the commissars of the CDC starts pontificating about ‘the science,’ one is probably similarly smitten by the stupidity that’s in the air.
Earlier I mentioned the sociologist David Riesman and his distinction between ‘inner-directed’ and ‘other-directed’ people. It takes no genius to realise that, to navigate one’s course through life relatively unscathed by peddlers of corruption, it is preferable to take one’s bearings from ‘inner direction’ by a set of values which promotes honesty and eschews mendacity, than from the ‘direction by others.’ Under present circumstances such other-directedness applies to the maze of lies and misinformation emanating from various government agencies as well as from certain peer groups, which today mostly comprise the vociferously self-righteous purveyors of the mainstream version of events. Inner-directness in the above sense, when constantly renewed, could be an effective guardian against stupidity.
Recall that Stiegler warned against the ‘deep malaise’ at contemporary universities in the context of what he called an ‘addictogenic’ society – that is, a society that engenders addictions of various kinds. Judging by the popularity of the video platform TikTok at schools and colleges, its use had already reached addiction levels by 2019, which raises the question, whether it should be appropriated by teachers as a ‘teaching tool,’ or whether it should, as some people think, be outlawed completely in the classroom.
Recall that, as an instance of video technology, TikTok is an exemplary embodiment of the pharmakon, and that, as Stiegler has emphasised, stupidity is the law of the pharmakon, which is, in turn, the law of knowledge. This is a somewhat confusing way of saying that knowledge and stupidity cannot be separated; where knowledge is encountered, its other, stupidity, lurks in the shadows.
Reflecting on the last sentence, above, it is not difficult to realise that, parallel to Freud’s insight concerning Eros and Thanatos, it is humanly impossible for knowledge to overcome stupidity once and for all. At certain times the one will appear to be dominant, while on different occasions the reverse will apply. Judging by the fight between knowledge and stupidity today, the latter ostensibly still has the upper hand, but as more people are awakening to the titanic struggle between the two, knowledge is in the ascendant. It is up to us to tip the scales in its favour – as long as we realise that it is a never-ending battle.
Government
“I Can’t Even Save”: Americans Are Getting Absolutely Crushed Under Enormous Debt Load
"I Can’t Even Save": Americans Are Getting Absolutely Crushed Under Enormous Debt Load
While Joe Biden insists that Americans are doing great…
Share this:


While Joe Biden insists that Americans are doing great - suggesting in his State of the Union Address last week that "our economy is the envy of the world," Americans are being absolutely crushed by inflation (which the Biden admin blames on 'shrinkflation' and 'corporate greed'), and of course - crippling debt.
The signs are obvious. Last week we noted that banks' charge-offs are accelerating, and are now above pre-pandemic levels.
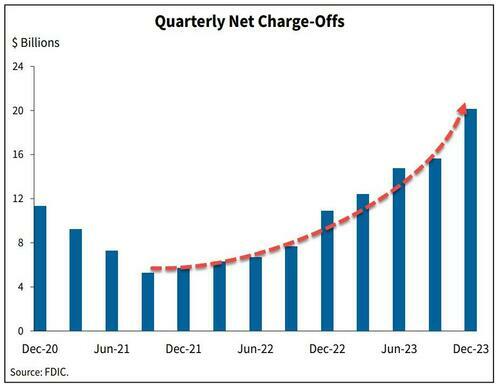
...and leading this increase are credit card loans - with delinquencies that haven't been this high since Q3 2011.
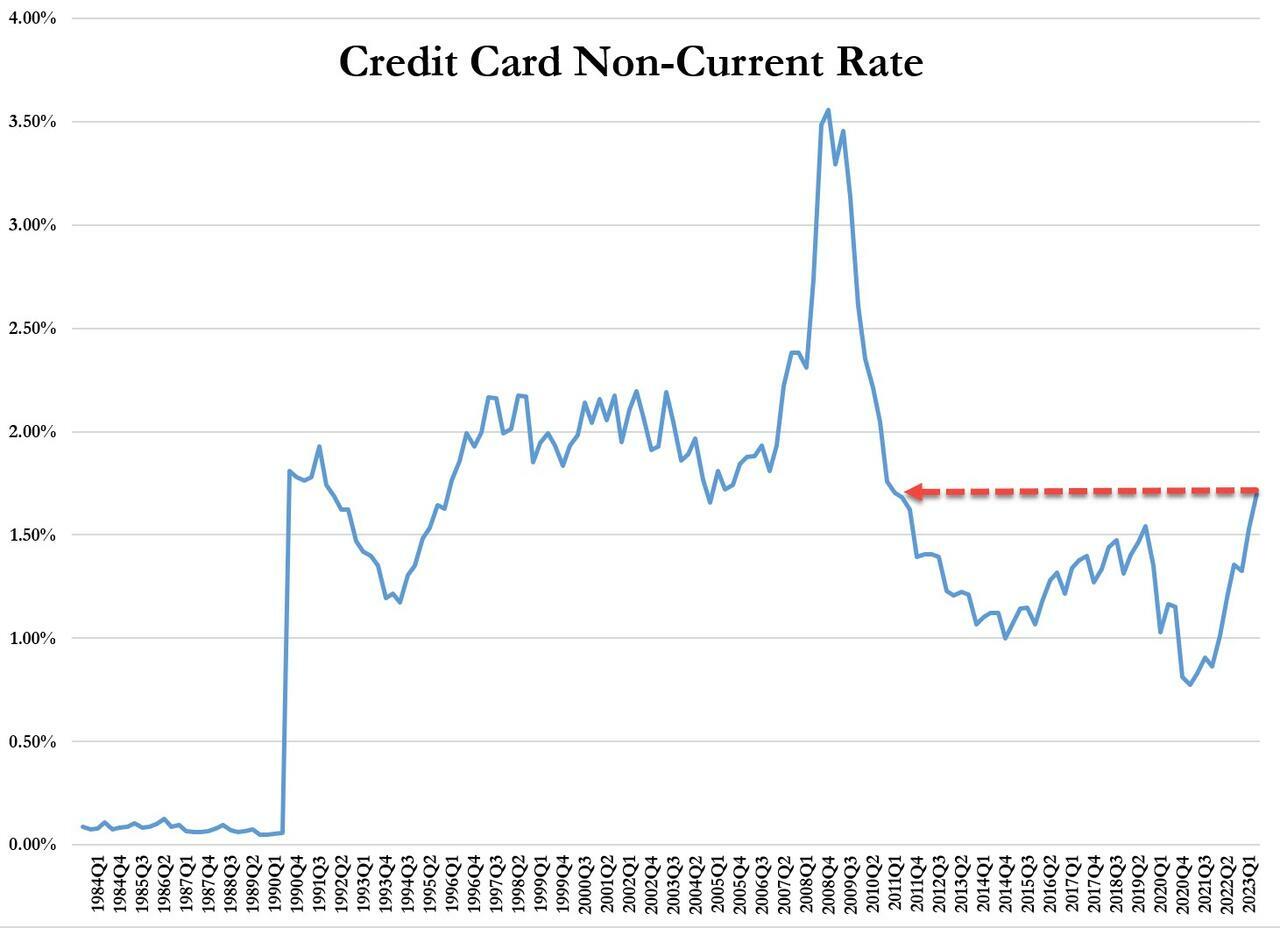
On top of that, while credit cards and nonfarm, nonresidential commercial real estate loans drove the quarterly increase in the noncurrent rate, residential mortgages drove the quarterly increase in the share of loans 30-89 days past due.
And while Biden and crew can spin all they want, an average of polls from RealClear Politics shows that just 40% of people approve of Biden's handling of the economy.
Crushed
On Friday, Bloomberg dug deeper into the effects of Biden's "envious" economy on Americans - specifically, how massive debt loads (credit cards and auto loans especially) are absolutely crushing people.
Two years after the Federal Reserve began hiking interest rates to tame prices, delinquency rates on credit cards and auto loans are the highest in more than a decade. For the first time on record, interest payments on those and other non-mortgage debts are as big a financial burden for US households as mortgage interest payments.
According to the report, this presents a difficult reality for millions of consumers who drive the US economy - "The era of high borrowing costs — however necessary to slow price increases — has a sting of its own that many families may feel for years to come, especially the ones that haven’t locked in cheap home loans."
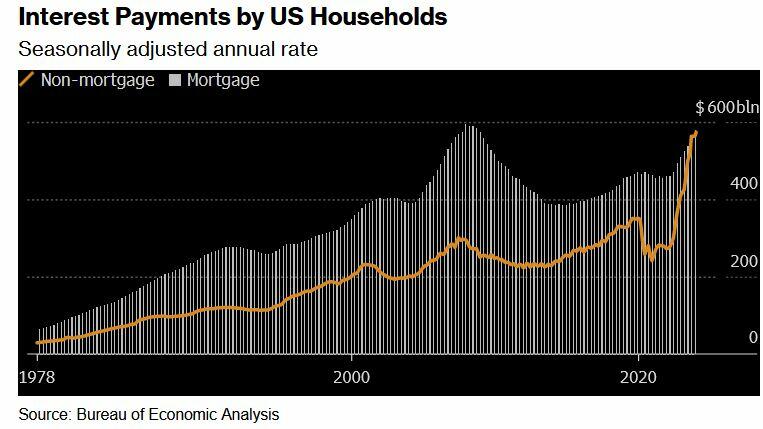
The Fed, meanwhile, doesn't appear poised to cut rates until later this year.
According to a February paper from IMF and Harvard, the recent high cost of borrowing - something which isn't reflected in inflation figures, is at the heart of lackluster consumer sentiment despite inflation having moderated and a job market which has recovered (thanks to job gains almost entirely enjoyed by immigrants).
In short, the debt burden has made life under President Biden a constant struggle throughout America.
"I’m making the most money I've ever made, and I’m still living paycheck to paycheck," 40-year-old Denver resident Nikki Cimino told Bloomberg. Cimino is carrying a monthly mortgage of $1,650, and has $4,000 in credit card debt following a 2020 divorce.

"There's this wild disconnect between what people are experiencing and what economists are experiencing."
CBS: Do you attribute the inflation crisis to the pandemic or Biden?
— RNC Research (@RNCResearch) March 15, 2024
WISCONSIN VOTER: "It's been YEARS now since the pandemic — I'm not buying that anymore. At first I did; I'm not buying that anymore because yogurt is STILL going up in price!" pic.twitter.com/apahb65scB
What's more, according to Wells Fargo, families have taken on debt at a comparatively fast rate - no doubt to sustain the same lifestyle as low rates and pandemic-era stimmies provided. In fact, it only took four years for households to set a record new debt level after paying down borrowings in 2021 when interest rates were near zero.
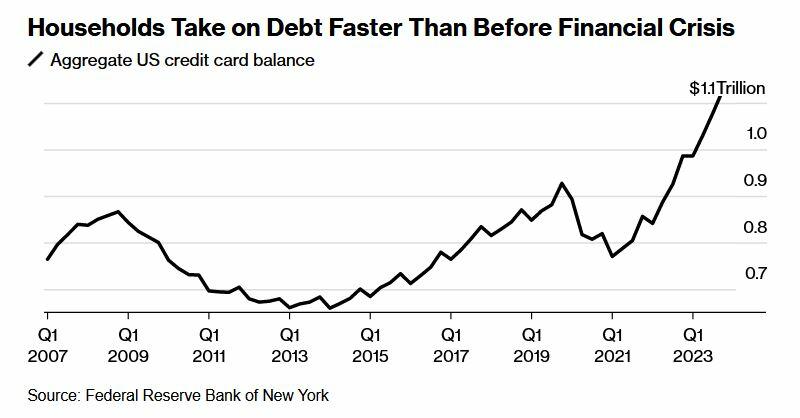
Meanwhile, that increased debt load is exacerbated by credit card interest rates that have climbed to a record 22%, according to the Fed.
[P]art of the reason some Americans were able to take on a substantial load of non-mortgage debt is because they’d locked in home loans at ultra-low rates, leaving room on their balance sheets for other types of borrowing. The effective rate of interest on US mortgage debt was just 3.8% at the end of last year.
Yet the loans and interest payments can be a significant strain that shapes families’ spending choices. -Bloomberg
And of course, the highest-interest debt (credit cards) is hurting lower-income households the most, as tends to be the case.
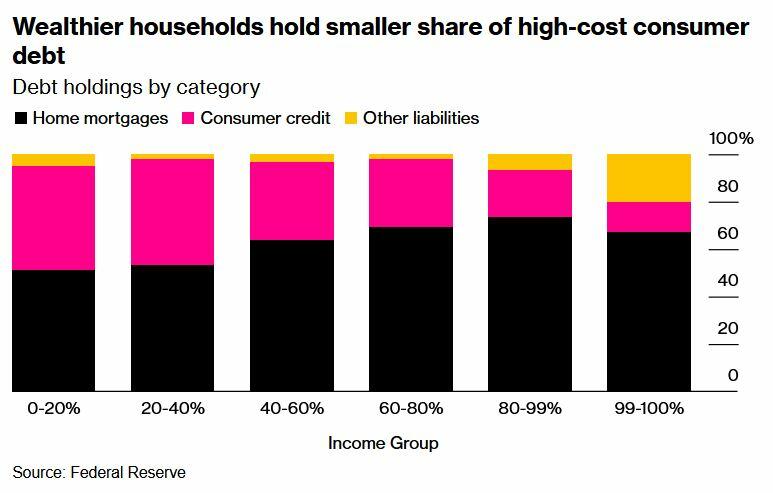
The lowest earners also understandably had the biggest increase in credit card delinquencies.

"Many consumers are levered to the hilt — maxed out on debt and barely keeping their heads above water," Allan Schweitzer, a portfolio manager at credit-focused investment firm Beach Point Capital Management told Bloomberg. "They can dog paddle, if you will, but any uptick in unemployment or worsening of the economy could drive a pretty significant spike in defaults."
"We had more money when Trump was president," said Denise Nierzwicki, 69. She and her 72-year-old husband Paul have around $20,000 in debt spread across multiple cards - all of which have interest rates above 20%.

Photographer: Jon Cherry/Bloomberg
During the pandemic, Denise lost her job and a business deal for a bar they owned in their hometown of Lexington, Kentucky. While they applied for Social Security to ease the pain, Denise is now working 50 hours a week at a restaurant. Despite this, they're barely scraping enough money together to service their debt.
The couple blames Biden for what they see as a gloomy economy and plans to vote for the Republican candidate in November. Denise routinely voted for Democrats up until about 2010, when she grew dissatisfied with Barack Obama’s economic stances, she said. Now, she supports Donald Trump because he lowered taxes and because of his policies on immigration. -Bloomberg
Meanwhile there's student loans - which are not able to be discharged in bankruptcy.
"I can't even save, I don't have a savings account," said 29-year-old in Columbus, Ohio resident Brittany Walling - who has around $80,000 in federal student loans, $20,000 in private debt from her undergraduate and graduate degrees, and $6,000 in credit card debt she accumulated over a six-month stretch in 2022 while she was unemployed.
"I just know that a lot of people are struggling, and things need to change," she told the outlet.
The only silver lining of note, according to Bloomberg, is that broad wage gains resulting in large paychecks has made it easier for people to throw money at credit card bills.
Yet, according to Wells Fargo economist Shannon Grein, "As rates rose in 2023, we avoided a slowdown due to spending that was very much tied to easy access to credit ... Now, credit has become harder to come by and more expensive."
According to Grein, the change has posed "a significant headwind to consumption."
Then there's the election
"Maybe the Fed is done hiking, but as long as rates stay on hold, you still have a passive tightening effect flowing down to the consumer and being exerted on the economy," she continued. "Those household dynamics are going to be a factor in the election this year."
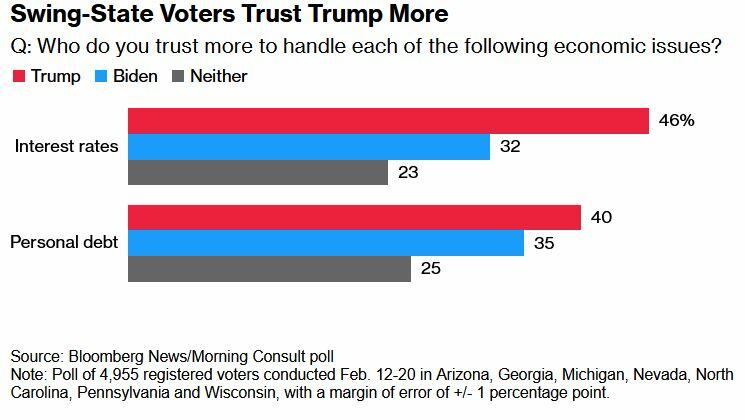
Meanwhile, swing-state voters in a February Bloomberg/Morning Consult poll said they trust Trump more than Biden on interest rates and personal debt.
Reverberations
These 'headwinds' have M3 Partners' Moshin Meghji concerned.
"Any tightening there immediately hits the top line of companies," he said, noting that for heavily indebted companies that took on debt during years of easy borrowing, "there's no easy fix."
International
Copper Soars, Iron Ore Tumbles As Goldman Says “Copper’s Time Is Now”
Copper Soars, Iron Ore Tumbles As Goldman Says "Copper’s Time Is Now"
After languishing for the past two years in a tight range despite recurring…
Share this:

{kind=link}
{kind=link}
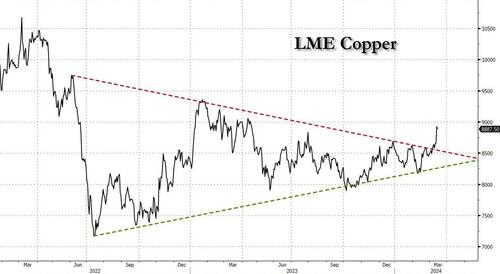
After languishing for the past two years in a tight range despite recurring speculation about declining global supply, copper has finally broken out, surging to the highest price in the past year, just shy of $9,000 a ton as supply cuts hit the market; At the same time the price of the world's "other" most important mined commodity has diverged, as iron ore has tumbled amid growing demand headwinds out of China's comatose housing sector where not even ghost cities are being built any more.
{kind=link}
Copper surged almost 5% this week, ending a months-long spell of inertia, as investors focused on risks to supply at various global mines and smelters. As Bloomberg adds, traders also warmed to the idea that the worst of a global downturn is in the past, particularly for metals like copper that are increasingly used in electric vehicles and renewables.
Yet the commodity crash of recent years is hardly over, as signs of the headwinds in traditional industrial sectors are still all too obvious in the iron ore market, where futures fell below $100 a ton for the first time in seven months on Friday as investors bet that China’s years-long property crisis will run through 2024, keeping a lid on demand.
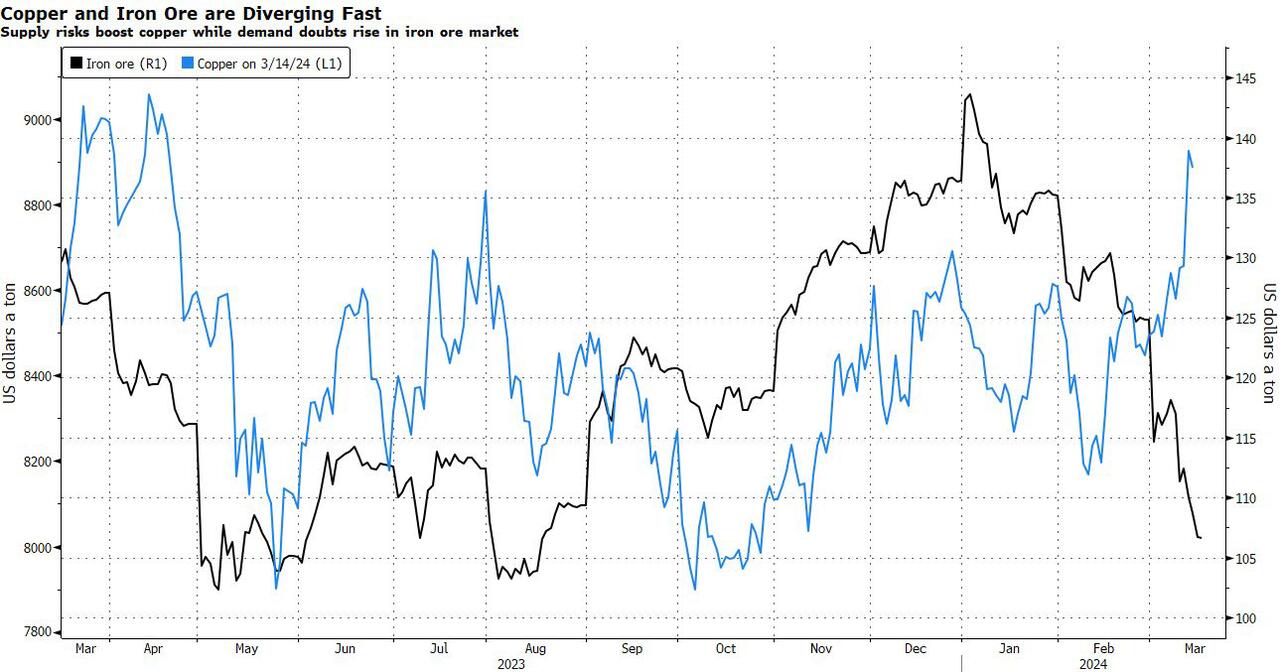
Indeed, while the mood surrounding copper has turned almost euphoric, sentiment on iron ore has soured since the conclusion of the latest National People’s Congress in Beijing, where the CCP set a 5% goal for economic growth, but offered few new measures that would boost infrastructure or other construction-intensive sectors.
As a result, the main steelmaking ingredient has shed more than 30% since early January as hopes of a meaningful revival in construction activity faded. Loss-making steel mills are buying less ore, and stockpiles are piling up at Chinese ports. The latest drop will embolden those who believe that the effects of President Xi Jinping’s property crackdown still have significant room to run, and that last year’s rally in iron ore may have been a false dawn.
Meanwhile, as Bloomberg notes, on Friday there were fresh signs that weakness in China’s industrial economy is hitting the copper market too, with stockpiles tracked by the Shanghai Futures Exchange surging to the highest level since the early days of the pandemic. The hope is that headwinds in traditional industrial areas will be offset by an ongoing surge in usage in electric vehicles and renewables.
And while industrial conditions in Europe and the US also look soft, there’s growing optimism about copper usage in India, where rising investment has helped fuel blowout growth rates of more than 8% — making it the fastest-growing major economy.
In any case, with the demand side of the equation still questionable, the main catalyst behind copper’s powerful rally is an unexpected tightening in global mine supplies, driven mainly by last year’s closure of a giant mine in Panama (discussed here), but there are also growing worries about output in Zambia, which is facing an El Niño-induced power crisis.
On Wednesday, copper prices jumped on huge volumes after smelters in China held a crisis meeting on how to cope with a sharp drop in processing fees following disruptions to supplies of mined ore. The group stopped short of coordinated production cuts, but pledged to re-arrange maintenance work, reduce runs and delay the startup of new projects. In the coming weeks investors will be watching Shanghai exchange inventories closely to gauge both the strength of demand and the extent of any capacity curtailments.
“The increase in SHFE stockpiles has been bigger than we’d anticipated, but we expect to see them coming down over the next few weeks,” Colin Hamilton, managing director for commodities research at BMO Capital Markets, said by phone. “If the pace of the inventory builds doesn’t start to slow, investors will start to question whether smelters are actually cutting and whether the impact of weak construction activity is starting to weigh more heavily on the market.”
* * *
Few have been as happy with the recent surge in copper prices as Goldman's commodity team, where copper has long been a preferred trade (even if it may have cost the former team head Jeff Currie his job due to his unbridled enthusiasm for copper in the past two years which saw many hedge fund clients suffer major losses).
As Goldman's Nicholas Snowdon writes in a note titled "Copper's time is now" (available to pro subscribers in the usual place)...

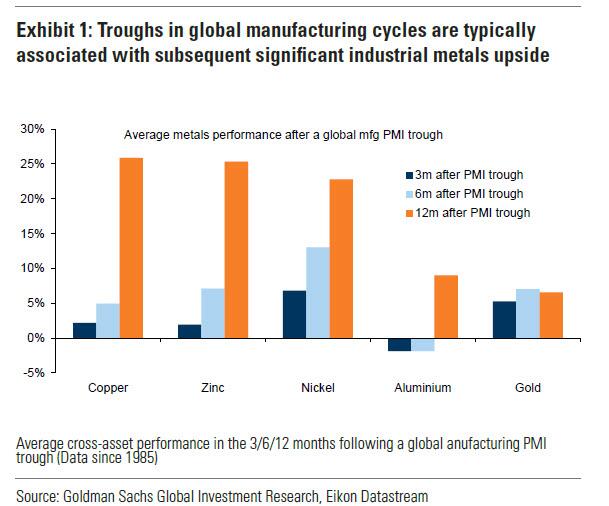
... there has been a "turn in the industrial cycle." Specifically according to the Goldman analyst, after a prolonged downturn, "incremental evidence now points to a bottoming out in the industrial cycle, with the global manufacturing PMI in expansion for the first time since September 2022." As a result, Goldman now expects copper to rise to $10,000/t by year-end and then $12,000/t by end of Q1-25.’
Here are the details:
Previous inflexions in global manufacturing cycles have been associated with subsequent sustained industrial metals upside, with copper and aluminium rising on average 25% and 9% over the next 12 months. Whilst seasonal surpluses have so far limited a tightening alignment at a micro level, we expect deficit inflexions to play out from quarter end, particularly for metals with severe supply binds. Supplemented by the influence of anticipated Fed easing ahead in a non-recessionary growth setting, another historically positive performance factor for metals, this should support further upside ahead with copper the headline act in this regard.
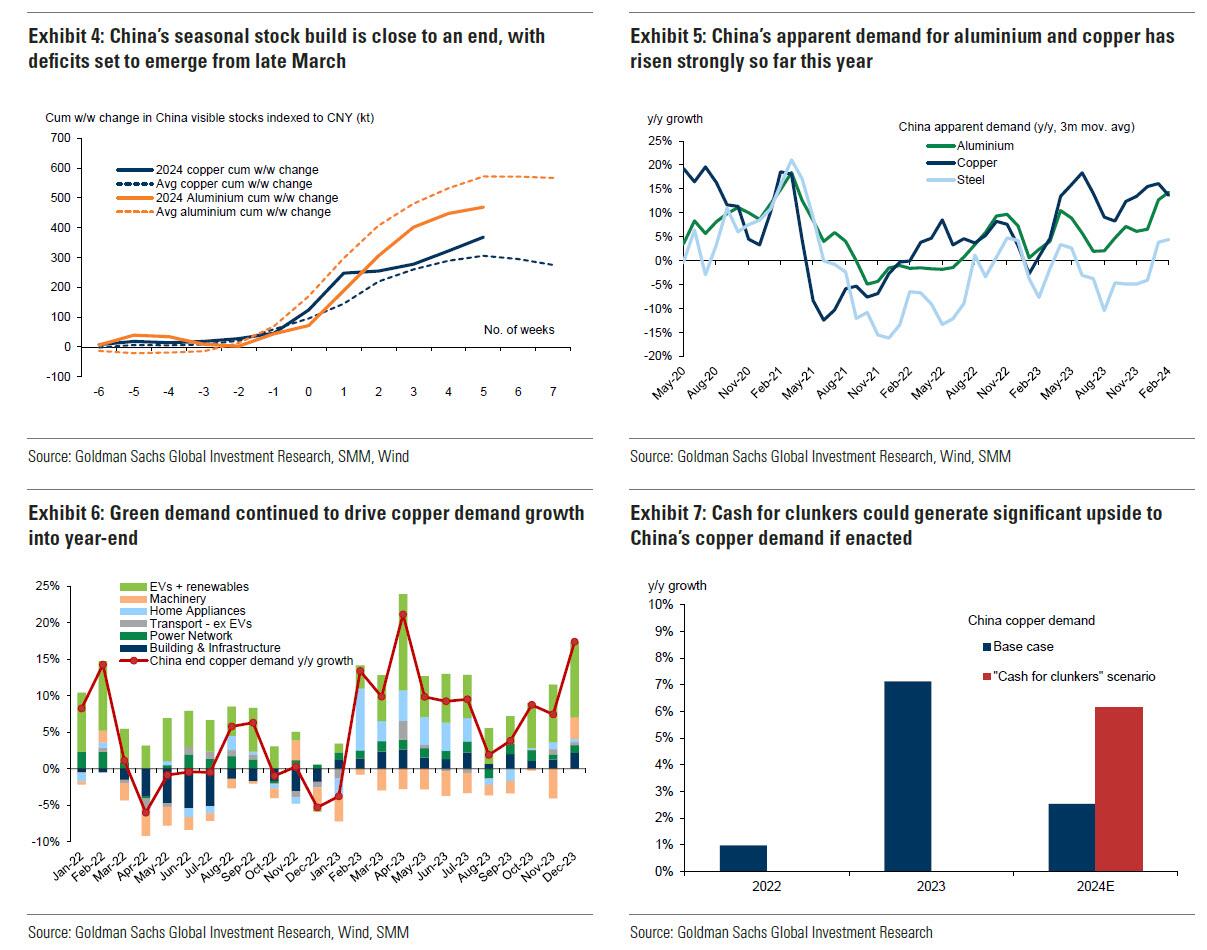
Goldman then turns to what it calls China's "green policy put":
Much of the recent focus on the “Two Sessions” event centred on the lack of significant broad stimulus, and in particular the limited property support. In our view it would be wrong – just as in 2022 and 2023 – to assume that this will result in weak onshore metals demand. Beijing’s emphasis on rapid growth in the metals intensive green economy, as an offset to property declines, continues to act as a policy put for green metals demand. After last year’s strong trends, evidence year-to-date is again supportive with aluminium and copper apparent demand rising 17% and 12% y/y respectively. Moreover, the potential for a ‘cash for clunkers’ initiative could provide meaningful right tail risk to that healthy demand base case. Yet there are also clear metal losers in this divergent policy setting, with ongoing pressure on property related steel demand generating recent sharp iron ore downside.
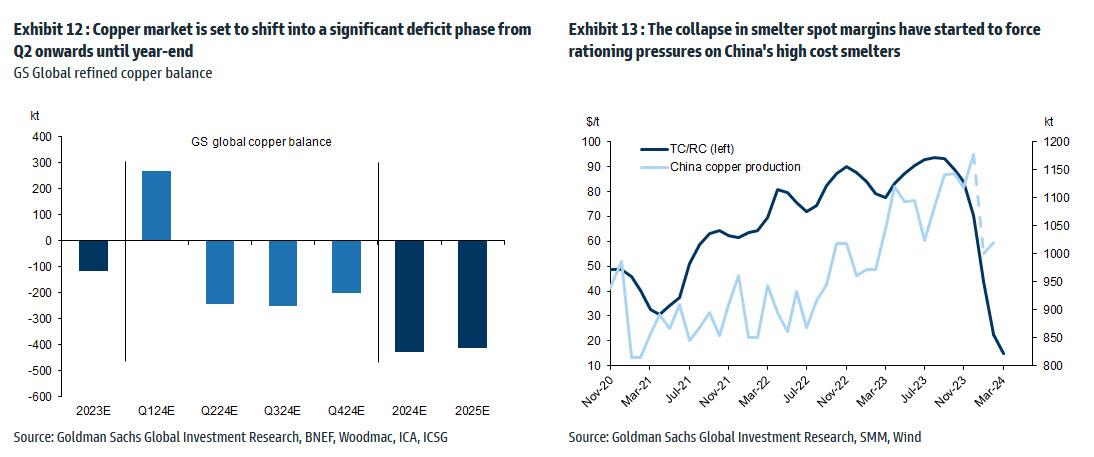
Meanwhile, Snowdon believes that the driver behind Goldman's long-running bullish view on copper - a global supply shock - continues:
Copper’s supply shock progresses. The metal with most significant upside potential is copper, in our view. The supply shock which began with aggressive concentrate destocking and then sharp mine supply downgrades last year, has now advanced to an increasing bind on metal production, as reflected in this week's China smelter supply rationing signal. With continued positive momentum in China's copper demand, a healthy refined import trend should generate a substantial ex-China refined deficit this year. With LME stocks having halved from Q4 peak, China’s imminent seasonal demand inflection should accelerate a path into extreme tightness by H2. Structural supply underinvestment, best reflected in peak mine supply we expect next year, implies that demand destruction will need to be the persistent solver on scarcity, an effect requiring substantially higher pricing than current, in our view. In this context, we maintain our view that the copper price will surge into next year (GSe 2025 $15,000/t average), expecting copper to rise to $10,000/t by year-end and then $12,000/t by end of Q1-25’
Another reason why Goldman is doubling down on its bullish copper outlook: gold.
The sharp rally in gold price since the beginning of March has ended the period of consolidation that had been present since late December. Whilst the initial catalyst for the break higher came from a (gold) supportive turn in US data and real rates, the move has been significantly amplified by short term systematic buying, which suggests less sticky upside. In this context, we expect gold to consolidate for now, with our economists near term view on rates and the dollar suggesting limited near-term catalysts for further upside momentum. Yet, a substantive retracement lower will also likely be limited by resilience in physical buying channels. Nonetheless, in the midterm we continue to hold a constructive view on gold underpinned by persistent strength in EM demand as well as eventual Fed easing, which should crucially reactivate the largely for now dormant ETF buying channel. In this context, we increase our average gold price forecast for 2024 from $2,090/toz to $2,180/toz, targeting a move to $2,300/toz by year-end.
Much more in the full Goldman note available to pro subs.


The War Between Knowledge And Stupidity


For-profit nursing homes are cutting corners on safety and draining resources with financial shenanigans − especially at midsize chains that dodge public scrutiny


Problems After COVID-19 Vaccination More Prevalent Among Naturally Immune: Study


“I Can’t Even Save”: Americans Are Getting Absolutely Crushed Under Enormous Debt Load


Looking Back At COVID’s Authoritarian Regimes


Trump nearly derailed democracy once − here’s what to watch out for in reelection campaign


Stock Market Today: Stocks turn lower as factory inflation spikes, retail sales miss target


Moderna turns the spotlight on long Covid with new initiatives


Copper Soars, Iron Ore Tumbles As Goldman Says “Copper’s Time Is Now”


COVID-19 vaccines: CDC says people ages 65 and up should get a shot this spring – a geriatrician explains why it’s vitally important
-

 Uncategorized3 weeks ago
Uncategorized3 weeks agoAll Of The Elements Are In Place For An Economic Crisis Of Staggering Proportions
-

 International1 week ago
International1 week agoEyePoint poaches medical chief from Apellis; Sandoz CFO, longtime BioNTech exec to retire
-

 Uncategorized4 weeks ago
Uncategorized4 weeks agoCalifornia Counties Could Be Forced To Pay $300 Million To Cover COVID-Era Program
-

 Uncategorized3 weeks ago
Uncategorized3 weeks agoApparel Retailer Express Moving Toward Bankruptcy
-

 Uncategorized4 weeks ago
Uncategorized4 weeks agoIndustrial Production Decreased 0.1% in January
-

 International1 week ago
International1 week agoWalmart launches clever answer to Target’s new membership program
-

 Spread & Containment3 days ago
Spread & Containment3 days agoIFM’s Hat Trick and Reflections On Option-To-Buy M&A
-

 Uncategorized4 weeks ago
Uncategorized4 weeks agoRFK Jr: The Wuhan Cover-Up & The Rise Of The Biowarfare-Industrial Complex