Healthy Nevada Project’s community-based approach reveals up to 90% of CDC Tier 1 genetic condition risks missed using clinical care guidelines
Credit: Healthy Nevada Project
Reno, Nev. (July 27, 2020) – In a new study published today in the journal Nature Medicine, researchers behind the Healthy Nevada Project® suggest that community-based genetic screening has the potential to efficiently identify individuals who may be at increased risk for three common inherited genetic conditions known to cause several forms of cancer and increased risk for heart disease or stroke.
In 2018, the Healthy Nevada Project® (the largest, community-based population health study combining genetic, clinical, environmental and social data) started notifying consenting study participants who have certain genetic variants which predispose them to the Centers for Disease Control and Prevention (CDC) Tier 1 genetic conditions. The study focused on identifying carriers of these conditions, which include Hereditary Breast and Ovarian Cancer, Lynch Syndrome, and Familial Hypercholesterolemia, because they are the most common conditions and early detection and treatment could significantly lower morbidity and mortality.
Initial results from almost 27,000 study participants showed that 90% of carriers of the CDC Tier 1 genetic conditions were not previously identified in a clinical setting. The authors conclude that population genetic screening would identify at-risk carriers not identified during routine care.
“Our first goal was to deliver actionable health data back to the participants of the study and understand whether or not broad population screening of CDC Tier 1 genomic conditions was a practical tool to identify at-risk individuals,” explained Joseph Grzymski, Ph.D., the principal investigator of the Healthy Nevada Project®, a research professor at the Desert Research Institute (DRI), chief scientific officer for Renown Health and lead author of the study.
“Now, two years into doing that, it is clear that the clinical guidelines for detecting risk in individuals are too narrow and miss too many at risk individuals.”
Within the group of 26,906 Healthy Nevada Project® participants that Grzymski’s research team studied, 358 (1.33%) were carriers for CDC Tier 1 conditions. However, only 25% of those individuals met clinical guidelines for genetic screening. Additionally, more than 20% of the carriers already had a
diagnosis of disease relevant to their underlying genetic condition.
“We’re at a point now where it’s possible to do clinical-grade genetic screening at population-scale,” added James Lu, M.D. Ph.D., co-founder and chief scientific officer of Helix and senior co-author of the study. “What this study demonstrates is the potential impact of doing so. By making genetic screening available more broadly, we can help the millions of Americans who are unaware that they are living at increased risk for highly actionable, genetic conditions take action.”
Most notably, the study found that of the 273 participants who were carriers of the CDC Tier 1 genetic conditions and had clinical record information, only 22 individuals showed any previous suspicion of their underlying genetic conditions.
“For the first time, we are providing information at the individual level so study participants can make lifesaving changes to reduce their risk based on their genetics,” said Anthony Slonim, M.D., Dr.PH., FACHE, president and CEO of Renown Health and co-director of the Project® study. “We’re conducting research on the community level to develop leading-edge research on health determinants for entire neighborhoods, states and eventually, the country. Returning these results allows us to understand the prevalence of genetically programmed diseases and illnesses that we have here in Nevada and ensure we are providing the best prevention and care plans. For the individual, the return of results can be lifechanging.”
According to the CDC, early detection and intervention of the Tier 1 genetic conditions could have a meaningful potential for clinical actionability and a positive impact on public health.
The Healthy Nevada Project®, which launched in 2016, offers free genetic testing to every Nevadan, aged 18 and older, interested in learning more about their health and genetic profile. With more than 50,000 study participants enrolled in four years, the Healthy Nevada Project® has become the fastest-enrolling
genetic study in the world.
###
For more about the Healthy Nevada Project® please visit healthynv.org
Renown Institute for Health Innovation is a collaboration between Renown Health – a locally governed and locally owned, not-for-profit integrated healthcare network serving Nevada, Lake Tahoe and northeast California; and the Desert Research Institute – a recognized world leader in investigating the effects of natural and humaninduced environmental change and advancing technologies aimed at assessing a changing planet. Renown IHI research teams are focused on integrating personal healthcare and environmental data with socioeconomic determinants to help Nevada address some of its most complex environmental health problems; while simultaneously expanding the state’s access to leading-edge clinical trials and fostering new connections with
biotechnology and pharmaceutical companies. Learn more at Healthynv.org.
Helix is the leading population genomics company operating at the intersection
of clinical care, research, and genomics. Its end-to-end platform enables health
systems, life sciences companies, and payers to advance genomic research and accelerate the integration of genomic data into clinical care. Powered by one of the world’s largest CLIA / CAP next-generation sequencing labs and its proprietary Exome+? assay, Helix supports all aspects of population genomics including recruitment and engagement, clinically actionable disease screening, return of results, and basic and translational research. In response to the COVID-19 public health crisis, Helix has launched a sensitive and scalable end-to-end COVID-19 test system to meet the needs of health systems, employers,
governments, and other organizations across the country. Learn more at http://www.helix.com.
Media Contact Justin Broglio justin.broglio@dri.edu
Chronic stress and inflammation linked to societal and environmental impacts in new study
From anxiety about the state of the world to ongoing waves of Covid-19, the stresses we face can seem relentless and even overwhelming. Worse, these stressors…
From anxiety about the state of the world to ongoing waves of Covid-19, the stresses we face can seem relentless and even overwhelming. Worse, these stressors can cause chronic inflammation in our bodies. Chronic inflammation is linked to serious conditions such as cardiovascular disease and cancer – and may also affect our thinking and behavior.
Credit: Image: Vodovotz et al/Frontiers
From anxiety about the state of the world to ongoing waves of Covid-19, the stresses we face can seem relentless and even overwhelming. Worse, these stressors can cause chronic inflammation in our bodies. Chronic inflammation is linked to serious conditions such as cardiovascular disease and cancer – and may also affect our thinking and behavior.
A new hypothesis published in Frontiers in Science suggests the negative impacts may extend far further.
“We propose that stress, inflammation, and consequently impaired cognition in individuals can scale up to communities and populations,” explained lead author Prof Yoram Vodovotz of the University of Pittsburgh, USA.
“This could affect the decision-making and behavior of entire societies, impair our cognitive ability to address complex issues like climate change, social unrest, and infectious disease – and ultimately lead to a self-sustaining cycle of societal dysfunction and environmental degradation,” he added.
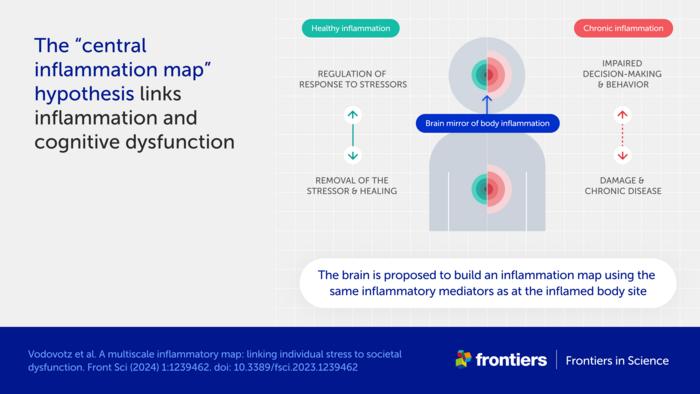
Bodily inflammation ‘mapped’ in the brain
One central premise to the hypothesis is an association between chronic inflammation and cognitive dysfunction.
“The cause of this well-known phenomenon is not currently known,” said Vodovotz. “We propose a mechanism, which we call the ‘central inflammation map’.”
The authors’ novel idea is that the brain creates its own copy of bodily inflammation. Normally, this inflammation map allows the brain to manage the inflammatory response and promote healing.
When inflammation is high or chronic, however, the response goes awry and can damage healthy tissues and organs. The authors suggest the inflammation map could similarly harm the brain and impair cognition, emotion, and behavior.
Accelerated spread of stress and inflammation online
A second premise is the spread of chronic inflammation from individuals to populations.
“While inflammation is not contagious per se, it could still spread via the transmission of stress among people,” explained Vodovotz.
The authors further suggest that stress is being transmitted faster than ever before, through social media and other digital communications.
“People are constantly bombarded with high levels of distressing information, be it the news, negative online comments, or a feeling of inadequacy when viewing social media feeds,” said Vodovotz. “We hypothesize that this new dimension of human experience, from which it is difficult to escape, is driving stress, chronic inflammation, and cognitive impairment across global societies.”
Inflammation as a driver of social and planetary disruption
These ideas shift our view of inflammation as a biological process restricted to an individual. Instead, the authors see it as a multiscale process linking molecular, cellular, and physiological interactions in each of us to altered decision-making and behavior in populations – and ultimately to large-scale societal and environmental impacts.
“Stress-impaired judgment could explain the chaotic and counter-intuitive responses of large parts of the global population to stressful events such as climate change and the Covid-19 pandemic,” explained Vodovotz.
“An inability to address these and other stressors may propagate a self-fulfilling sense of pervasive danger, causing further stress, inflammation, and impaired cognition in a runaway, positive feedback loop,” he added.
The fact that current levels of global stress have not led to widespread societal disorder could indicate an equally strong stabilizing effect from “controllers” such as trust in laws, science, and multinational organizations like the United Nations.
“However, societal norms and institutions are increasingly being questioned, at times rightly so as relics of a foregone era,” said Prof Paul Verschure of Radboud University, the Netherlands, and a co-author of the article. “The challenge today is how we can ward off a new adversarial era of instability due to global stress caused by a multi-scale combination of geopolitical fragmentation, conflicts, and ecological collapse amplified by existential angst, cognitive overload, and runaway disinformation.”
Reducing social media exposure as part of the solution
The authors developed a mathematical model to test their ideas and explore ways to reduce stress and build resilience.
“Preliminary results highlight the need for interventions at multiple levels and scales,” commented co-author Prof Julia Arciero of Indiana University, USA.
“While anti-inflammatory drugs are sometimes used to treat medical conditions associated with inflammation, we do not believe these are the whole answer for individuals,” said Dr David Katz, co-author and a specialist in preventive and lifestyle medicine based in the US. “Lifestyle changes such as healthy nutrition, exercise, and reducing exposure to stressful online content could also be important.”
“The dawning new era of precision and personalized therapeutics could also offer enormous potential,” he added.
At the societal level, the authors suggest creating calm public spaces and providing education on the norms and institutions that keep our societies stable and functioning.
“While our ‘inflammation map’ hypothesis and corresponding mathematical model are a start, a coordinated and interdisciplinary research effort is needed to define interventions that would improve the lives of individuals and the resilience of communities to stress. We hope our article stimulates scientists around the world to take up this challenge,” Vodovotz concluded.
The article is part of the Frontiers in Science multimedia article hub ‘A multiscale map of inflammatory stress’. The hub features a video, an explainer, a version of the article written for kids, and an editorial, viewpoints, and policy outlook from other eminent experts: Prof David Almeida (Penn State University, USA), Prof Pietro Ghezzi (University of Urbino Carlo Bo, Italy), and Dr Ioannis P Androulakis (Rutgers, The State University of New Jersey, USA).
Journal
Frontiers in Science
DOI
10.3389/1239462
Method of Research
Computational simulation/modeling
Subject of Research
Not applicable
Article Title
A multiscale inflammatory map: linking individual stress to societal dysfunction
Article Publication Date
12-Mar-2024
COI Statement
YV is a cofounder of, and stakeholder in, Immunetrics, Inc. and a consultant to Anuna AI. PFMJV is the founder of, and stakeholder in, Eodyne Systems s.l. and Sapiens5 Holding BV. DLK was employed by Tangelo – Intend, Inc. Neither these companies nor the funders mentioned above were involved in the study design, data collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication. The companies mentioned above also did not provide funding for the study. The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The authors YV and PV declared that they are editorial board members of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Acadia’s Nuplazid fails PhIII study due to higher-than-expected placebo effect
After years of trying to expand the market territory for Nuplazid, Acadia Pharmaceuticals might have hit a dead end, with a Phase III fail in schizophrenia…
After years of trying to expand the market territory for Nuplazid, Acadia Pharmaceuticals might have hit a dead end, with a Phase III fail in schizophrenia due to the placebo arm performing better than expected.
Steve Davis
“We will continue to analyze these data with our scientific advisors, but we do not intend to conduct any further clinical trials with pimavanserin,” CEO Steve Davis said in a Monday press release. Acadia’s stock $ACAD dropped by 17.41% before the market opened Tuesday.
Pimavanserin, a serotonin inverse agonist and also a 5-HT2A receptor antagonist, is already in the market with the brand name Nuplazid for Parkinson’s disease psychosis. Efforts to expand into other indications such as Alzheimer’s-related psychosis and major depression have been unsuccessful, and previous trials in schizophrenia have yielded mixed data at best. Its February presentation does not list other pimavanserin studies in progress.
The Phase III ADVANCE-2 trial investigated 34 mg pimavanserin versus placebo in 454 patients who have negative symptoms of schizophrenia. The study used the negative symptom assessment-16 (NSA-16) total score as a primary endpoint and followed participants up to week 26. Study participants have control of positive symptoms due to antipsychotic therapies.
The company said that the change from baseline in this measure for the treatment arm was similar between the Phase II ADVANCE-1 study and ADVANCE-2 at -11.6 and -11.8, respectively. However, the placebo was higher in ADVANCE-2 at -11.1, when this was -8.5 in ADVANCE-1. The p-value in ADVANCE-2 was 0.4825.
In July last year, another Phase III schizophrenia trial — by Sumitomo and Otsuka — also reported negative results due to what the company noted as Covid-19 induced placebo effect.
According to Mizuho Securities analysts, ADVANCE-2 data were disappointing considering the company applied what it learned from ADVANCE-1, such as recruiting patients outside the US to alleviate a high placebo effect. The Phase III recruited participants in Argentina and Europe.
Analysts at Cowen added that the placebo effect has been a “notorious headwind” in US-based trials, which appears to “now extend” to ex-US studies. But they also noted ADVANCE-1 reported a “modest effect” from the drug anyway.
Nonetheless, pimavanserin’s safety profile in the late-stage study “was consistent with previous clinical trials,” with the drug having an adverse event rate of 30.4% versus 40.3% with placebo, the company said. Back in 2018, even with the FDA approval for Parkinson’s psychosis, there was an intense spotlight on Nuplazid’s safety profile.
Acadia previously aimed to get Nuplazid approved for Alzheimer’s-related psychosis but had many hurdles. The drug faced an adcomm in June 2022 that voted 9-3 noting that the drug is unlikely to be effective in this setting, culminating in a CRL a few months later.
As for the company’s next R&D milestones, Mizuho analysts said it won’t be anytime soon: There is the Phase III study for ACP-101 in Prader-Willi syndrome with data expected late next year and a Phase II trial for ACP-204 in Alzheimer’s disease psychosis with results anticipated in 2026.
Acadia collected $549.2 million in full-year 2023 revenues for Nuplazid, with $143.9 million in the fourth quarter.
"Beware the Ides of March,” Shakespeare quotes the soothsayer’s warning Julius Caesar about what turned out to be an impending assassination on March 15. The death of American liberty happened around the same time four years ago, when the orders went out from all levels of government to close all indoor and outdoor venues where people gather.
It was not quite a law and it was never voted on by anyone. Seemingly out of nowhere, people who the public had largely ignored, the public health bureaucrats, all united to tell the executives in charge – mayors, governors, and the president – that the only way to deal with a respiratory virus was to scrap freedom and the Bill of Rights.
And they did, not only in the US but all over the world.
The forced closures in the US began on March 6 when the mayor of Austin, Texas, announced the shutdown of the technology and arts festival South by Southwest. Hundreds of thousands of contracts, of attendees and vendors, were instantly scrapped. The mayor said he was acting on the advice of his health experts and they in turn pointed to the CDC, which in turn pointed to the World Health Organization, which in turn pointed to member states and so on.
There was no record of Covid in Austin, Texas, that day but they were sure they were doing their part to stop the spread. It was the first deployment of the “Zero Covid” strategy that became, for a time, official US policy, just as in China.
It was never clear precisely who to blame or who would take responsibility, legal or otherwise.
This Friday evening press conference in Austin was just the beginning. By the next Thursday evening, the lockdown mania reached a full crescendo. Donald Trump went on nationwide television to announce that everything was under control but that he was stopping all travel in and out of US borders, from Europe, the UK, Australia, and New Zealand. American citizens would need to return by Monday or be stuck.
Americans abroad panicked while spending on tickets home and crowded into international airports with waits up to 8 hours standing shoulder to shoulder. It was the first clear sign: there would be no consistency in the deployment of these edicts.
There is no historical record of any American president ever issuing global travel restrictions like this without a declaration of war. Until then, and since the age of travel began, every American had taken it for granted that he could buy a ticket and board a plane. That was no longer possible. Very quickly it became even difficult to travel state to state, as most states eventually implemented a two-week quarantine rule.
The next day, Friday March 13, Broadway closed and New York City began to empty out as any residents who could went to summer homes or out of state.
On that day, the Trump administration declared the national emergency by invoking the Stafford Act which triggers new powers and resources to the Federal Emergency Management Administration.
In addition, the Department of Health and Human Services issued a classified document, only to be released to the public months later. The document initiated the lockdowns. It still does not exist on any government website.
The White House Coronavirus Response Task Force, led by the Vice President, will coordinate a whole-of-government approach, including governors, state and local officials, and members of Congress, to develop the best options for the safety, well-being, and health of the American people. HHS is the LFA [Lead Federal Agency] for coordinating the federal response to COVID-19.
Closures were guaranteed:
Recommend significantly limiting public gatherings and cancellation of almost all sporting events, performances, and public and private meetings that cannot be convened by phone. Consider school closures. Issue widespread ‘stay at home’ directives for public and private organizations, with nearly 100% telework for some, although critical public services and infrastructure may need to retain skeleton crews. Law enforcement could shift to focus more on crime prevention, as routine monitoring of storefronts could be important.
In this vision of turnkey totalitarian control of society, the vaccine was pre-approved: “Partner with pharmaceutical industry to produce anti-virals and vaccine.”
The National Security Council was put in charge of policy making. The CDC was just the marketing operation. That’s why it felt like martial law. Without using those words, that’s what was being declared. It even urged information management, with censorship strongly implied.
The timing here is fascinating. This document came out on a Friday. But according to every autobiographical account – from Mike Pence and Scott Gottlieb to Deborah Birx and Jared Kushner – the gathered team did not meet with Trump himself until the weekend of the 14th and 15th, Saturday and Sunday.
According to their account, this was his first real encounter with the urge that he lock down the whole country. He reluctantly agreed to 15 days to flatten the curve. He announced this on Monday the 16th with the famous line: “All public and private venues where people gather should be closed.”
This makes no sense. The decision had already been made and all enabling documents were already in circulation.
There are only two possibilities.
One: the Department of Homeland Security issued this March 13 HHS document without Trump’s knowledge or authority. That seems unlikely.
Two: Kushner, Birx, Pence, and Gottlieb are lying. They decided on a story and they are sticking to it.
Trump himself has never explained the timeline or precisely when he decided to greenlight the lockdowns. To this day, he avoids the issue beyond his constant claim that he doesn’t get enough credit for his handling of the pandemic.
With Nixon, the famous question was always what did he know and when did he know it? When it comes to Trump and insofar as concerns Covid lockdowns – unlike the fake allegations of collusion with Russia – we have no investigations. To this day, no one in the corporate media seems even slightly interested in why, how, or when human rights got abolished by bureaucratic edict.
As part of the lockdowns, the Cybersecurity and Infrastructure Security Agency, which was and is part of the Department of Homeland Security, as set up in 2018, broke the entire American labor force into essential and nonessential.
They also set up and enforced censorship protocols, which is why it seemed like so few objected. In addition, CISA was tasked with overseeing mail-in ballots.
Only 8 days into the 15, Trump announced that he wanted to open the country by Easter, which was on April 12. His announcement on March 24 was treated as outrageous and irresponsible by the national press but keep in mind: Easter would already take us beyond the initial two-week lockdown. What seemed to be an opening was an extension of closing.
This announcement by Trump encouraged Birx and Fauci to ask for an additional 30 days of lockdown, which Trump granted. Even on April 23, Trump told Georgia and Florida, which had made noises about reopening, that “It’s too soon.” He publicly fought with the governor of Georgia, who was first to open his state.
Before the 15 days was over, Congress passed and the president signed the 880-page CARES Act, which authorized the distribution of $2 trillion to states, businesses, and individuals, thus guaranteeing that lockdowns would continue for the duration.
There was never a stated exit plan beyond Birx’s public statements that she wanted zero cases of Covid in the country. That was never going to happen. It is very likely that the virus had already been circulating in the US and Canada from October 2019. A famous seroprevalence study by Jay Bhattacharya came out in May 2020 discerning that infections and immunity were already widespread in the California county they examined.
What that implied was two crucial points: there was zero hope for the Zero Covid mission and this pandemic would end as they all did, through endemicity via exposure, not from a vaccine as such. That was certainly not the message that was being broadcast from Washington. The growing sense at the time was that we all had to sit tight and just wait for the inoculation on which pharmaceutical companies were working.
By summer 2020, you recall what happened. A restless generation of kids fed up with this stay-at-home nonsense seized on the opportunity to protest racial injustice in the killing of George Floyd. Public health officials approved of these gatherings – unlike protests against lockdowns – on grounds that racism was a virus even more serious than Covid. Some of these protests got out of hand and became violent and destructive.
Meanwhile, substance abuse rage – the liquor and weed stores never closed – and immune systems were being degraded by lack of normal exposure, exactly as the Bakersfield doctors had predicted. Millions of small businesses had closed. The learning losses from school closures were mounting, as it turned out that Zoom school was near worthless.
It was about this time that Trump seemed to figure out – thanks to the wise council of Dr. Scott Atlas – that he had been played and started urging states to reopen. But it was strange: he seemed to be less in the position of being a president in charge and more of a public pundit, Tweeting out his wishes until his account was banned. He was unable to put the worms back in the can that he had approved opening.
By that time, and by all accounts, Trump was convinced that the whole effort was a mistake, that he had been trolled into wrecking the country he promised to make great. It was too late. Mail-in ballots had been widely approved, the country was in shambles, the media and public health bureaucrats were ruling the airwaves, and his final months of the campaign failed even to come to grips with the reality on the ground.
At the time, many people had predicted that once Biden took office and the vaccine was released, Covid would be declared to have been beaten. But that didn’t happen and mainly for one reason: resistance to the vaccine was more intense than anyone had predicted. The Biden administration attempted to impose mandates on the entire US workforce. Thanks to a Supreme Court ruling, that effort was thwarted but not before HR departments around the country had already implemented them.
As the months rolled on – and four major cities closed all public accommodations to the unvaccinated, who were being demonized for prolonging the pandemic – it became clear that the vaccine could not and would not stop infection or transmission, which means that this shot could not be classified as a public health benefit. Even as a private benefit, the evidence was mixed. Any protection it provided was short-lived and reports of vaccine injury began to mount. Even now, we cannot gain full clarity on the scale of the problem because essential data and documentation remains classified.
After four years, we find ourselves in a strange position. We still do not know precisely what unfolded in mid-March 2020: who made what decisions, when, and why. There has been no serious attempt at any high level to provide a clear accounting much less assign blame.
Not even Tucker Carlson, who reportedly played a crucial role in getting Trump to panic over the virus, will tell us the source of his own information or what his source told him. There have been a series of valuable hearings in the House and Senate but they have received little to no press attention, and none have focus on the lockdown orders themselves.
The prevailing attitude in public life is just to forget the whole thing. And yet we live now in a country very different from the one we inhabited five years ago. Our media is captured. Social media is widely censored in violation of the First Amendment, a problem being taken up by the Supreme Court this month with no certainty of the outcome. The administrative state that seized control has not given up power. Crime has been normalized. Art and music institutions are on the rocks. Public trust in all official institutions is at rock bottom. We don’t even know if we can trust the elections anymore.
In the early days of lockdown, Henry Kissinger warned that if the mitigation plan does not go well, the world will find itself set “on fire.” He died in 2023. Meanwhile, the world is indeed on fire. The essential struggle in every country on earth today concerns the battle between the authority and power of permanent administration apparatus of the state – the very one that took total control in lockdowns – and the enlightenment ideal of a government that is responsible to the will of the people and the moral demand for freedom and rights.
How this struggle turns out is the essential story of our times.
CODA: I’m embedding a copy of PanCAP Adapted, as annotated by Debbie Lerman. You might need to download the whole thing to see the annotations. If you can help with research, please do.
We use cookies on our website to give you the most relevant experience by remembering your preferences and repeat visits. By clicking “Accept”, you consent to the use of ALL the cookies.
This website uses cookies to improve your experience while you navigate through the website. Out of these, the cookies that are categorized as necessary are stored on your browser as they are essential for the working of basic functionalities of the website. We also use third-party cookies that help us analyze and understand how you use this website. These cookies will be stored in your browser only with your consent. You also have the option to opt-out of these cookies. But opting out of some of these cookies may affect your browsing experience.
Necessary cookies are absolutely essential for the website to function properly. These cookies ensure basic functionalities and security features of the website, anonymously.
Cookie
Duration
Description
cookielawinfo-checbox-analytics
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Analytics".
cookielawinfo-checbox-functional
11 months
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Functional".
cookielawinfo-checbox-others
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Other.
cookielawinfo-checkbox-necessary
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookies is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-performance
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Performance".
viewed_cookie_policy
11 months
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
Functional cookies help to perform certain functionalities like sharing the content of the website on social media platforms, collect feedbacks, and other third-party features.
Performance cookies are used to understand and analyze the key performance indexes of the website which helps in delivering a better user experience for the visitors.
Analytical cookies are used to understand how visitors interact with the website. These cookies help provide information on metrics the number of visitors, bounce rate, traffic source, etc.
Advertisement cookies are used to provide visitors with relevant ads and marketing campaigns. These cookies track visitors across websites and collect information to provide customized ads.













































{kind=link}